|
http://www.fda.gov/ohrms/dockets/ac/05/transcripts/2005-4172t1.htm
FDA Hearing- CHAIRMAN
EDMISTON: If
the devices
(jetguns) are
used in a
compliant manner
the way they're
meant to be
used, do you
think the
devices are
safe?
DR. FRIEDE: The
devices that we
have seen
without a
protection cap,
we have data
from the calves
and the data
from the Hoffman
study in Brazil
to show that
frequent
contamination of
the ejected did
take place. And
that
contamination
was clearly of a
level of blood
that we are
convinced can
carry disease.
So the devices
which do not
have a
protection cap
which are to be
used for
giving
intramuscular
injection we are
convinced that
these carry a
significant
risk.
U.S. FOOD
AND DRUG
ADMINISTRATION
GENERAL HOSPITAL
AND PERSONAL USE
DEVICES PANEL
OF THE MEDICAL
DEVICES ADVISORY
COMMITTEE
THIRTY-FIFTH
MEETING
TUESDAY, AUGUST
9, 2005
The
above‑entitled
matter met in
Salons A, B, and
C of the Hilton
Washington, D.C.
North, 620 Perry
Parkway,
Gaithersburg,
Maryland, at
8:00 a.m.,
Charles E.
Edmiston, Jr.,
Ph.D.,
Chairperson,
presiding.
PRESENT:
CHARLES E.
EDMISTON, JR.,
Ph.D,
Chairperson
MATTHEW J.
ARDUINO, D.Phil,
Voting Member
RICHARD O.
BUTCHER, M.D.,
Voting Member
YARDIN B. DAVID,
Ed.D., Voting
Member
BONNIE M. WORD,
M.D., Voting
Member
TERRY LAYTON,
Ph.D., Industry
Representative
CAROLYN N.
PETERSEN, M.S.,
Consumer
Representative
CHIU S. LIN,
Ph.D., Director,
Division of
Anesthesiology,
General
Hospital,
Infection
Control, and
Dental Devices
SCOTT A.
COLBURN, BSN,
RN, LT, USPHS
Executive
Secretary
FDA
PRESENTERS:
THOMAS GROSS,
M.D., M.P.H.,
Director,
Division of
Postmarket
Surveillance,
Office of
Surveillance and
Biometrics
SHEILA MURPHEY,
M.D., Chief,
Infection
Control
Devices Branch
ANTHONY D.
WATSON, M.S.,
M.B.A., Chief,
General Hospital
Devices Branch
JASON F. LIPMAN,
Lead Reviewer,
General Hospital
Devices Branch
SHEWIT BEZABEH,
M.D., M.P.H.,
Medical Officer,
Division of
Anesthesiology,
General
Hospital,
Infection
Control, and
Dental
Devices
DAYAWANSA G.
RANAMUKHA-ARACHCHI,
Ph.D., Molecular
Biologist/Genomics,
Office of
Science and
Laboratories,
Division of
Biology
INVITED GUEST
PRESENTER:
MARTIN FRIEDE,
Ph.D.,
Initiative for
Vaccine
Research, World
Health
Organization
INDUSTRY
PRESENTERS:
DARIN LEE
ZEHRUNG, Program
for Appropriate
Technology in
Health (PATH)
MARK KANE,
Program for
Appropriate
Technology in
Health (PATH)
LINDA D'ANTONIO,
D'Antonio
Consultants
International
KATHLEEN
CALLENDER,
Genesis Medical
Technologies
PUBLIC SPEAKER:
HARRY HOOKS
HCVets.com
A-G-E-N-D-A
INTRODUCTIONS...................................
5
Executive
Secretary
Colburn...............
6
CONDITION OF
APPROVAL
STUDIES: RECENT
CHANGES IN CDRH
Dr.
Gross................................
13
DIVISION/BRANCH
UPDATE
Dr. Lin, DAGID
Division
Director.........
21
Dr. Murphey,
Chief Infection
Control
Devices
Branch.....................
23
Mr. Watson,
Chief, General
Hospital
Devices
Branch.....................
27
PUBLIC HEARING
SESSION
Harry Hooks,
HCVets.com..................
38
PRESENTATIONS BY
FDA
Introduction and
welcome,
Mr.
Watson.........................
50
Mr. Lipman...............................
51
Dr. Bezabeh..............................
59
Dr.
Ranamukha-arachchi...................
72
Questions by
Members to FDA
presenters... 86
PRESENTATIONS BY
CDC AND WHO
Dr. Friede,
WHO.........................
102
PRESENTATIONS BY
INDUSTRY
Dr. Zehrung,
PATH.......................
134
Dr. Kane,
PATH..........................
154
Ms. D'Antonio,
DCI......................
179
Ms. Callender,
Genesis Medical
Technologies......................
180
PANEL
DELIBERATIONS...........................
181
OPEN PUBLIC
HEARING 246
Dr. Kane,
PATH..........................
246
P-R-O-C-E-E-D-I-N-G-S
8:06 a.m.
CHAIRMAN
EDMISTON: Good
morning. I'd
like to welcome
to the 35th
meeting of the
General Hospital
and Personal Use
Device Panel.
I also want to
request everyone
in attendance at
this meeting to
sign in on the
attendance sheet
that is
available on the
table outside
the door.
I will note for
the record the
voting members
present
constitute a
quorum as
defined by 21
CRF Part 14.
At this time I
would like each
panel member at
the table to
introduce him or
herself and
state his or her
specialty
position, title,
institution and
status on the
Panel. And I'll
start with my
left, Dr. Lin.
DR. LIN: Hi.
Good morning. My
name is Chiu
Lin. I'm the
Director of
Division of
Anesthesiology,
General
Hospital,
Infection
Control and
Dental Device in
FDA.
MS. PETERSEN: My
name is Carolyn
Petersen. I'm a
web editor at
Mayo Clinic in
Rochester,
Minnesota. And
I'm here as the
consumer
representative.
MR. DAVID: Good
morning. My name
is Yardin David.
I'm Director of
Biomedical
Engineering
Department at
Texas Children's
Hospital in
Houston and
Assistant
Professor at
Baylor College
of Medicine,
Department of
Pediatrics.
EXECUTIVE
SECRETARY
COLBURN: Good
morning. My name
is Lieutenant
Scott Colburn. I
am the Executive
Secretary to the
General Hospital
and Personal Use
Devices Panel.
CHAIRMAN
EDMISTON: My
name is Charles
Edmiston. I am a
faculty member
at the Medical
College of
Wisconsin and
hospital
epidemiologist.
DR. WORD: Hi. My
name is Bonnie
Word. I am on
faculty at
Baylor College
of Medicine also
at Texas
Children's
Medical Center
where I'm the
Chief of the
infectious
disease clinic
and travel
medicine
clinics.
DR. ARDUINO: Hi.
My name is Matt
Arduino, and I'm
the lead
microbiologist
in the
epidemiology and
laboratory
branch at the
Division of
Health Care
Quality and
Promotion at the
Center for
Disease Control
and Prevention.
DR. BUTCHER: I'm
Richard Butcher,
a physician a
San Diego,
general practice
with Care View
Medical Group.
DR. LAYTON: Good
morning. I'm
Terry Layton, a
biomedical
engineer. I'm
industry
representative
on this Panel.
And I'm from
Laytech,
Incorporated out
of Chicago,
Illinois.
CHAIRMAN
EDMISTON: Thank
you.
Lieutenant Scott
Colburn, the
Executive
Secretary, would
like to make
some
introductory
remarks.
Lt. Colburn?
EXECUTIVE
SECRETARY
COLBURN: Before
I start the
remarks, I'd
like to
introduce Ms.
Mary Ann Killian
from the Ethics
Integrity staff
to read the
conflict of
interest
statement for
the members of
the Panel.
MS. KILLIAN:
Thank you.
The Food and
Drug
Administration
is convening
today's meeting
of the General
Hospital And
Personal Use
Devices Panel of
the Medical
Device Advisory
Committee under
the authority of
the Federal
Advisory Act of
1972. The
Advisory Panel
meeting provides
transparency
into the
Agency's
deliberative
processes. With
the exception of
the industry
representative,
all members of
the Panel are
special
government
employees or
regular federal
employees from
other agencies
and are subject
to the Federal
Conflict of
Interest laws
and regulations.
Consequently, in
the interest of
transparency and
the spirit of
disclosure, the
following
information on
the status of
this Advisory
Committee
Panel's
compliance with
the Federal
Ethics and
Conflict of
Interest laws
covered by but
not limited to
those found at
18 USC 208 and
21 USC 355(N)(4)
is being
provided to the
participants in
today's meeting
and to the
public.
FDA has
determined that
members and
consultants of
this Panel are
in compliance
with Federal
Ethics and
Conflict of
Interest laws.
Under 18 USC 208
Congress has
authorized FDA
to grant waivers
to special
government
employees who
have limited
financial
conflicts when
it is determined
that the
Agency's need
for a particular
individual's
service
outweighs his or
her potential
financial
conflict of
interest.
Members and
consultants who
are special
government
employees at
today's meeting
have been
screened for
potential
financial
conflicts of
interest of
their own as
well as those
imputed to them
including those
of their
employers,
spouse or minor
child related to
the discussion
of today's
meeting. These
interests may
include
investments,
consulting
expert witness
testimony,
contracts
grants, creative
teaching,
speaking,
writing,
patents,
royalties and
primary
employment.
Today's agenda
involves a
discussion on
methods to
assess the
potential of
disease
transmission by
multi-use nozzle
jet injectors;
that is jet
injectors for
which the fluid
path for the
injection is
used more than
once. The
discussion will
also include
premarket
testing,
recommendations
to address this
issue. This is a
general matters
meeting during
which the topic
of discussion is
limited to
recommendations
or
considerations
of broad
legislative
proposals,
regulatory
initiatives or
policy
developments
that affect an
industry, group
of manufacturers
or health care
providers. So
any conflict of
interest waivers
granted for this
meeting are
broad and
general in
nature.
A copy of the
written conflict
of interest
waiver statement
may be obtained
by writing to
the Agency's
Freedom of
Information
Office, 12A30 of
the Parklawn
Building.
Based on the
agenda for
today's meeting
and all
financial
interests by the
Panel
participants it
has been
determined that
all interests in
firms regulated
by the Center
for Devices and
Radiological
Health present
no actual or
appearance of
conflict of
interest for
today's meeting.
The following
Panel
participants
have not
received a
conflict of
interest waiver
to participate
in today's
meeting: Dr.
Charles Edmiston,
Dr. Matthew
Arduino, Dr.
Richard Butcher,
Dr. Bonnie Word,
Dr. Yardin David
and Ms. Carolyn
Petersen.
In addition, Dr.
Terry Layton has
been invited to
participate as
the industry rep
acting on behalf
of all related
industry, and is
employed by
Laytech,
Incorporated.
With regard to
FDA's guest
speakers, the
Agency has
determined that
the information
provided by
these speakers
is essential.
The following
interests are
being made
public to allow
the audience to
objectively
evaluate any
presentation
and/or comments
made by the
speakers:
Dr. Bruce
Weniger, who is
a guest speaker
with us today,
has acknowledged
that his
employer, the
Centers for
Disease Control
and Prevention,
has financial
interest in
firms at issue.
The financial
interests and
professional
relationships
are in the form
of research
contracts and
educational
projects
involving
multiple-use jet
injectors.
Dr. Martin
Friede, who is
also a guest
speaker with us
today, has
acknowledged
that his
employed the
World Health
Organization has
interest in
today's topic in
the form of
pending clinical
trials. As guest
speakers, these
individuals will
not participate
in Panel
deliberation.
Members and
consultants of
the Committee
are reminded
that if the work
of the Committee
moves from
matters of
general
applicability to
matters that are
more specific,
for example
product or firms
identified, the
FDA shall end
the discussion
promptly and
each special
government
employee's
financial
interest will be
reexamined in
relation to the
particular
matters so that
a determination
may be made on
whether
exclusion from
further
discussion is
required. All
exclusions will
be noted for the
record.
Finally, in the
interests of
public
transparency
with respect to
all other
participants, we
ask that they
publicly
disclose prior
to making any
remarks any
current or
previous
financial
involvement with
any firm whose
products they
may wish to
comment upon.
This statement
will be
available for
review at the
registration
table during
this meeting and
will be included
as part of the
official meeting
transcript.
Thank you.
EXECUTIVE
SECRETARY
COLBURN: Thank
you, Ms.
Killian.
The FDA seeks
communication
with industry
and the clinical
community in a
number of
different ways.
First, FDA
welcomes and
encourages
premeetings with
sponsors prior
to all IDE and
PMA submissions.
This affords the
sponsor an
opportunity to
discuss issues
that could
impact the
review process.
Second, the FDA
communicates
through the use
of guidance
documents.
Toward this end,
FDA develops two
types of
guidance
documents for
manufacturers to
follow in
submitting a
premarket
application. One
type is simply a
summary of the
information that
has historically
been requested
on devices that
are well
understood in
order to
determine
substantial
equivalence. The
second type of
guidance
document is one
that develops as
we learn about
new technology.
The FDA welcomes
and encourages
the Panel and
industry to
provide comments
concerning our
guidance
documents.
I'd also like to
remind you that
the tentative
dates for the
next meeting on
the General
Hospital and
Personal Use
Devices Panel is
scheduled for
September 27,
2005. You may
wish to pencil
in this date on
your calendar,
but please
recognize that
this date is
tentative at
this time.
The first item
on our agenda is
a presentation
by Dr. Tom Gross
from the Office
of Surveillance
and Biometrics.
He will discuss
the conditions
of approval
studies and
recent changes
in CDRH.
Dr. Gross?
DR. GROSS: Good
morning.
As was stated,
I'm Tom Gross.
I'm the Director
of the Division
of Postmarket
Surveillance in
our Office of
Surveillance and
Biometrics. And
I'd like to take
a few minutes of
your time today
to talk bout
recent changes
in our
conditions of
approval study
program.
Before I do
that, I'd like
to touch based
on some of the
essential
functions that
our office
serves for the
center. And
those are
presented in
this slide here.
First and
foremost, we
provide support
for premarket
review. We have
a large group of
statisticians
who address all
statistical
aspects of
premarket
submissions. We
also have a
group of
epidemiologists
who are involved
in PMA review
teams and help
design condition
of approval
studies.
We are also
responsible
through our
nationwide
passive
surveillance
systems to
detect signals
of potential
public health
problems. That's
our Medical
Device Reporting
system or MDR
system. And our
network of user
facilities
throughout the
United States
for our MedSun
network.
Thirdly, we're
responsible for
risk
characterization
and analysis of
these potential
public safety
issues. This is
done primarily
by our
epidemiology
staff doing
everything from
systematic
literature
reviews to de
novo studies.
We also
coordinate our
center response
on these public
health issues.
We convene
committees of
center experts
to deliberate
these issues and
to present their
recommendations
to center senior
staff for
action.
And lastly, we
have a staff who
interpret our
medical device
reporting
regulations;
what needs to be
reported under
what
circumstances,
and also to
follow-up on
violations of
those reporting
requirements.
Now let's turn
to our condition
of approval
study program.
As most of you
know, these
studies are
ordered as a
condition of
approval of our
PMA products.
And the
regulations
clearly
stipulate the
following:
That post
approval
requirements can
include
continuing
evaluation and
periodic
reporting on the
safety,
effectiveness
and reliability
of the device
for its intended
use. This
regulation gives
us our broad
authority in
ordering these
post approval
studies.
Next slide.
Now about the
middle of 2002
our office took
a snapshot of
the center's
activities with
regard to the
condition
approval study
program to see
how well the
center was
doing. And the
study basically
involved looking
at PMAs that
were approved
from 1998
through the year
2000. All
tolled, there
were 127 PMAs
that were
approved during
that period of
time. 45 of
those had
clinical
condition of
approval study
orders.
At the end of
the day what did
we find? That
CDRH had limited
procedures for
tracking study
progress for
results, that
our IT and other
systems were
wholly deficient
in this regard.
There's large
turnover of lead
reviewers that
resulted in lack
of follow-up. Up
to 40 percent of
individuals who
are lead
reviewers at the
time the PMA
came in the door
were no longer
associated with
that PMA when we
did this study.
And lastly,
there was lack
of premarket
resources. Those
resources were
devoted to
premarket
submissions and
there was little
left for
oversight of
condition
approval
studies.
Next slide.
So based on
these results
and based on an
ongoing pilot we
had of
epidemiologists
involved with
PMA reviews we
decided there
was need for a
change. And the
goal for that
change basically
focused on the
following:
To obtain
useful, timely
and quality
postmarket
information on
the safety and
effectiveness of
devices as they
move into the
marketplace;
To better
characterize the
risk and benefit
profile of these
devices. For
instances, their
long term
performance, and
to add to our
ability to make
sound scientific
decisions based
on these timely
and high quality
studies.
So what did we
do in terms of
change? The next
two slides
speaks to this.
We transferred
the condition of
approval study
program from our
premarket side
of the house,
the Office of
Device
Evaluation, to
our postmarket
side of the
house, the
Office of
Surveillance and
Biometrics. We
did that
effective
January of this
year.
We did that for
two reasons.
One, our office
has the
resources to
oversee the
program and we
also have the
resident
expertise in
epidemiologists
to be part of
this program.
We developed and
instituted an
automatic
tracking system
for these
studies so we
could
acknowledge
receipt of the
protocols and
interim study
reports, and
follow-up when
reports were not
received.
Next slide.
Most
importantly, we
added
epidemiologist
to all the PMA
review teams for
all the five
review divisions
within the
Office of Device
Evaluation. The
epidemiologists
were tasked with
the development
of postmarketing
monitoring plans
during the
premarket
review. These
plans spoke to
the best means
of monitoring
the safety of
these products
in the
postmarket
period.
Epidemiologists
assumed the lead
in developing
and formulating
postmarket
questions, the
lead in the
design of
condition
approval study
protocols and
tracking those
study results
over the period
of the study.
And throughout
this process we
collaborated
very closely
with all members
of the PMA
review team.
Next slide.
In addition, we
addressed
motivation for
study conduct,
meaning how best
can industry do
these studies
and how best can
FDA participate
in these
studies. And
first and
foremost,
obviously it's
important to
address the
important
postmarket
questions: What
are the
essential
questions that
need to be
addressed in
these condition
approval studies
and to develop a
good study
protocol to
address those
questions and
objectives.
We had to
acknowledge the
receipt of these
protocols and
study reports in
real time,
providing real
time feedback to
the industry.
As part of a
guidance
document we hope
to issue soon,
we hope to be
transparent with
regard to these
studies by
posting the
status of these
studies on
CDRH's website.
And lastly,
there are other
authorities that
we can levy if
companies do not
perform these
studies with due
diligence. And
those other
authorities give
us leeway in
terms of
misbanding the
product or
levying monetary
penalties if the
companies
continue to fail
to do those
studies.
Next slide.
And lastly,
what's the
impact on the
Advisory Panel?
We will attempt
to lay out the
important post
approval public
health questions
for the Panel's
deliberation and
possible
considerations.
And we will also
inform the
panel, that is
FDA and
industry, on a
periodic basis
about the
results of these
studies that
were approved.
Thank you very
much.
EXECUTIVE
SECRETARY
COLBURN: Thank
you, Dr. Gross.
Before I turn
the meeting back
over to Dr.
Edmiston, I'd
like to ask that
all cell phones
and pagers be
turned off or
placed in the
silent mode,
please, so they
do not interrupt
the business
during the time
of this meeting.
Dr. Edmiston?
CHAIRMAN
EDMISTON: Thank
you.
At this time we
have several
presentations
from
representatives
of the Division
of
Anesthesiology,
General Hospital
Infection
Control and
Dental Devices.
Our first
presenter will
be Mr. Lin,
Director of the
Division of
Anesthesiology,
General Hospital
Infection
Control and
Dental Devices.
He will provide
a very brief
update of the
Division's
activities.
Dr. Lin?
DR. LIN: Good
morning.
I thought I will
spend a few
minutes to talk
about what the
current update.
I know that
since the last
Panel meeting
the Division has
changed
significantly.
So I will spend
a few minutes to
talk about what
the Division,
and following my
presentation the
two branch
chiefs are going
to give you an
update what each
branch chief's
activities.
As you probably
may know, the
Center for
Device and
Radiological
Health composed
of at least six
office, and
because of the
time I don't
want to go into
the detail, but
next slide,
please.
Office of Device
Evaluation,
where that's
most of us work
in the Office of
Device
Evaluation, is
composed of five
divisions. And
division is
divide according
to product line
that we are
responsible for
reviewing. And
the divisions of
Anesthesia,
General Hospital
and Infection
Control and
Dental Device
are one of those
divisions in the
Office of Device
Evaluations.
Next one.
Currently the
Division has
myself is
Division
Director. And
then we have Dr.
Ginette Michaud
who is sitting
in the audience.
Dr. Michaud, can
you -- she's my
Deputy Director.
Next.
From the
Division's name
imply that we
are responsible
for four product
lines. One is
the
Anesthesiology
and Respiratory
Device branch.
And at current
the branch chief
is Ms. Ann
Graham. And some
of you probably
already met. We
have a panel
meeting not long
ago.
And then we have
a Dental Device
branch, and the
chair of the
branch is Dr.
Susan Runner.
Some probably
also met. We
also had panel
meeting a few
months ago.
And then the
General Hospital
Device branch is
headed by Mr.
Tony Watson. Is
right here.
And Infection
Control Device
branch is headed
by Dr. Sheila
Murphey. Is
right here.
Next. And the
FDA's, our
divisions for
your information
we have three
major panel
involved with
our product
lines. First one
is
Anesthesiology
and Respiratory
Device Panel.
And the second
one is Dental
Product Panel.
And the third
one is what we
are here now,
that's General
Hospital and
Personal Use
Devices Panel,
which is here by
General Hospital
Device branch
and Infection
Control Device
branch.
And Dr. Murphey
is going to give
you an update
what Infection
Control Devices
activity.
Thank you.
DR. MURPHEY:
Good morning.
I'm Dr. Sheila
Murphey, the
branch chief for
the Infection
Control Devices
Branch.
Next slide.
Our branch has a
number of
scientific
reviewers with
different
backgrounds. We
currently have
three
microbiologists,
that will be
four in two
weeks. We have
just filled the
open position
mentioned.
We have a
biochemist, a
nurse and a
biologist. We
also have a
fellow whom we
share with OSEL,
whom we will
have for another
two months.
My own
background is
clinical
infectious
disease and
hospital
infection
control.
Next slide.
Our branch
reviews a number
of devices. We
fall into two
major
categories. We
look at
everything
related to
sterilization.
All types of
sterilizers, the
medical washers,
washer
disinfectors and
endoscope washer
disinfectors.
We also review
high level
disinfectants
and liquid
sterilants.
We are
responsible for
looking at the
reprocessing of
single use
medical devices.
We look at the
sterilization
packaging
systems and the
indicators to
indicate the
adequacy of the
sterilization
process.
We also review
personal
protective
equipment;
gloves, gowns,
masks and such
devices.
We also are
responsible for
reviewing needle
disposal units
and needle
destruction
devices, which
are PMA devices.
Next slide,
please.
Recently
published
guidance
documents for
our branch
include the
Guidance for
Industry and FDA
Medical User Fee
and
Modernization
Act of 2002, The
Validation Data
in Premarket
Notification
Submissions For
Reprocessing
Single Use
Devices. This is
a preliminary
document. There
is work underway
for a final
guidance
document. Also
the Premarket
Approval
Applications for
Absorbable
Powders for
Lubricating a
Surgeon's Glove,
the Surgical
Mask Guidance,
the Submissions
for Chemical
Indicators
Guidance.
Next slide,
please.
We have several
new guidance
documents in
progress. The
one that we hope
will be
available soon
will be one
addressing
antimicrobial
agents on
medical devices.
We are working
on a guidance
document for the
reprocessing of
single use
medical devices
and also one for
standardizing
the reprocessing
of reusable
devices. This
will concentrate
particularly on
cleaning
devices.
We have a
guidance
document in
progress for the
germicides for
reprocessing
reusable
hemodialyzer
systems.
May I have the
next slide?
We are also
working on
revisions to
existing
guidance
documents, the
one that covers
surgical gowns
and drapes, the
one that address
chemotherapy
gloves, medical
sterilization
packaging
systems. Another
for needle
disposal devices
and biological
indicators.
Can I have the
next slide,
please?
Review
challenges for
our division
relate to the
technology that
we review.
Nontraditional
sterilization
technology is a
fascinating new
area. There's a
great deal of
new technology
coming along,
and validating
the processes
involved can be
challenging.
The reprocessing
of single use
medical devices
is progressing.
We are seeing
increasingly
complex devices
being submitted
for
reprocessing,
the validation
of this is a
very complex
process, as is
the need for
standardization
among the
entities
conducting the
reprocessing of
single use
medical devices.
And finally, the
cleaning of
medical devices,
a general topic
which addresses
not just single
use medical
devices but
really all
medical devices,
is something
that needs
validation and
more
standardization
we believe
throughout the
industry.
Thank you very
much.
EXECUTIVE
SECRETARY
COLBURN: Thank
you.
Next we have Mr.
Anthony Watson,
Chief of the
General Hospital
Devices Branch
who will give a
brief update on
the FDA General
Hospital Device
activities
related to this
Panel.
MR. WATSON: Good
morning. My name
is Anthony
Watson. As
mentioned, I am
the Chief of the
General Hospital
Devices Branch,
and I'm going to
give you an
update on what
has happened in
our branch since
the last Panel
meeting.
Just to give you
some idea of my
background, I'm
a general
engineer. I've
been with the
FDA for a little
over 11 years. I
was a reviewer
in another
branch for 10
years and I took
over this branch
in spring of
last year.
This Panel last
met August 2,
1999 and two
guidance were
discussed at
that Panel
meeting, one for
pen injectors
and one for jet
injectors.
Obviously, jet
injectors are
the topic of
today. In
particular,
during that
discussion six
years ago there
was quite a bit
of discussion
regarding cross
contamination of
jet injectors.
And that is
actually going
to be the focus
for today's
meeting.
As you might
imagine, in six
years there's
some degree of
turnover. This
branch has had a
significant
amount of
turnover. As I
mentioned, I
became the
branch chief
last year, March
of 2004. Our
branch right now
consists of
seven members
with varying
backgrounds. We
have three
nurses, three
engineers of
different
backgrounds,
different types.
And we have one
microbiologist.
We have in our
branch a lot of
devices that
have broad uses.
As our name
implies, General
Hospital, we
have general use
devices,
needle-free,
obviously jet
injectors as
we're going to
talk about them
today and well
as pen
injectors. We do
both implantable
and external
infusion pumps,
syringes and
needles and IV
admin sets, and
long term and
short term
intravascular
catheters.
In addition to
that, we also do
devices that
have sharpes
injury
protection
features. These
differ from the
devices that Dr.
Murphey's group
reviews in the
fact that they
deal with them
after they are
used, and these
devices actually
incorporate
sharps injury
protection
features.
And one area
that's really
growing for us
is the general
use medical
software area.
We're starting
to see a lot
more action in
this particular
area.
We also review
acupuncture
needles,
pharmacy
compounding
devices. And we
deal quite
heavily with
combination
products. Those
are products
that have
devices and
either a
combination of
biologics or
drugs.
We've also
published a
number of
guidance
documents. In
2001 we
published a
Class C Special
Control Guidance
document for
Pharmacy
Compounding
Systems. And
that was also
concordant with
the actual
classification
of those
products.
We put out a
guidance
document in 2002
for sharps
injury
prevention
features, which
we are
presenting in
the process of
updating.
And in 2004 we
cleared an
interesting
device,
implantable
radio frequency
transponder
system for
patient
identification,
health
information. And
in accordance
with that
process we also
generated a
Class II special
control guidance
document.
The last, the
most recent
guidance
document that
was published
was
intravascular
admin set. This
is a revision to
an existing
guidance
document. And
that was
published in
April of this
year.
We are in the
process, we have
quite a bit of
guidance
documents that
have been around
for a while. And
we are in the
process of
updating some
and actually
generating some
new guidance
documents.
The pen injector
and jet
injector, as I
mentioned
earlier, six
years ago we had
a discussion
about what kind
of information
would go into
those guidance
documents. We're
now going to be
actually
generating those
guidance
documents. And
we're going to
be revising our
guidance
documents to
infusion pumps,
intravascular
catheters and
pharmacy
compounding
devices.
We've had a
number of
clearances over
the years that
have some
interesting
issues and
features
associated with
them. But
perhaps the one
that's generated
the most
interest was
this implantable
radio frequency
transponder
system for
patient
identification
and health
information. And
it's significant
in a number of
ways.
First of all,
just to briefly
describe the
device, the
device really
consists of
three
components. A
chip that's
implanted in the
skin that's
about the size
of a grain of
rice, an
introducer which
is used to
implant the
device and a
reader. The
reader actually
-- the device
itself, the chip
only contains a
patient
identification
number. It
doesn't contain
any other
information
about the
patient. But the
reader can
extract that
code, then using
that code
whoever is
authorized to go
into a
proprietary
database can
then take that
information and
pull up the
patient's
information.
That health
information is
supplied by the
patient. It is
generated from
any other
location. So the
patient actually
gets to tell the
person what they
want the person
to know.
That device was
cleared under
the de novo
review process,
and it was
really -- I was
real proud of
the review team
because it was
really a cross
cutting kind of
product. We had
people that
looked at the
electromagnetic
compatibility of
the product, the
bio
compatibility of
the product. The
MRI
compatibility of
the product.
There was
software
discussions
about data
security, data
integrity.
And where's
Gail? Is she in
here? Am I
missing
anything, Gail?
I think I got it
all.
The bottom line
was it was under
a de novo review
process, which
is a process
that's beyond
the scope of me
describing it at
this Panel
meeting, but it
required us to
do all that
within 60 days
and generate a
Class II
guidance
document as
well. So I was
real proud of
the review team
for that. And
you may hear
more about this
product.
Next slide,
please.
We have a number
of challenges
that we're
facing in our
branch. And I
have combination
products up
there because
they're always a
challenge.
Inter-center
consults,
getting consults
with other
centers to
review them in
our statutory
time frames is
always a
challenge. Our
other centers
have been great
for helping us
with that, but
it is a very
difficult thing
to do.
Cross-labeling
of combination
products.
There's always a
question whether
the device
component should
reference the
drug or biologic
component and
vice versa, how
much of that
should occur.
We're always
dealing with
that.
And, as I
mentioned, the
growing area for
us is software
based devices.
One of the
things that
really is a
challenge is
that these
devices we're
talking about a
lot of times are
just software.
There is no
hardware
associated with
them. We're
talking code,
maybe put on a
CD, a DVD,
placed on a
server,
something like
that. And how do
you regulate
that? What
performance do
you look for? I
mean, what are
the issues
associated with
that?
And we're also
dealing with a
number of
existing devices
that have IT
technology
applied to them,
particularly in
the area of
wireless
communication
through
networks. And
where does the
device begin and
where does the
device end?
That's always a
question there.
But we are
seeing more
action in that
area.
Human factors:
This one is
basically
related to our
attempts to
address human
errors due to
human factors.
Particularly in
the area of
infusion pumps,
there's always a
question about
whether these
errors can be
prevented
through proper
human factors,
considerations
and the design
process. So
we're really
starting to
emphasize that
in our review
process. And
it's not just
infusion pumps,
it's really any
device that we
deal with that
has a high human
machine
interface. We
want to make
sure that we're
asking those
people to look
at those human
factors in the
review process
at the design
stage.
And one area
that's really
sort of exploded
for us recently
is the use of --
I have
peripheral
catheters up,
but we're also
talking central
catheters as
well that are
using power
injection for
contrast media.
Obviously these
type of
procedures
generate high
pressures, high
flow rates. A
lot of catheters
on the market
are not actually
tested to that
level. And we
want to make
sure that we've
got the proper
testing for
that. That's a
challenge
because these
devices are made
with different
materials,
different sizes.
No two are
alike,
basically. So
we're trying to
develop testing
for that, is
really a
challenge for
us. But we do
have some great
ground work. Our
reviewers have
done a good job
about
identifying the
clinical issues
and taking a
look at the
engineering
aspects.
And one other
aspect that we
are really
concerned about
is what
information do
we need to
provide for
users. It's
really critical
that the users
know how to
incorporate that
in the way
they're using
the products.
So that's the
General Hospital
Devices update.
And thank you
very much.
CHAIRMAN
EDMISTON: Thank
you.
We will now
proceed with the
first of our two
half hour open
public hearing
sessions. The
second open
public hearing
session will
follow the Panel
discussion this
afternoon.
During this
period public
attendees are
given an
opportunity to
address the
Panel to present
data or views
relevant to the
Panel's
activities. Some
individuals have
already given
advance notice
of wishing to
address the
Panel. Each
speaker will be
given a 15
minute
opportunity to
speak.
I would like to
remind the
public observers
at this time
that while this
portion of the
meeting is open
to public
observation,
public attendees
may not
participate
except at the
specific request
of the Chair.
We would also
ask at this time
that persons
addressing the
Panel come
forward, keeping
in mind this
presentation is
being
transcribed and
speak clearly
into the
microphone.
If you have a
hard copy of
your
presentation,
please provide
that to my
colleague,
Lieutenant
Colburn or leave
it on the
transcription
desk.
The following
statement is to
be read verbatim
at the general
matters meeting.
"Both the Food
and Drug
Administration
and the public
believe in a
transparent
process for
information
gathering and
decision making.
To ensure such
transparency at
the open public
hearing session
of the Advisory
Committee
meeting the FDA
believes that it
is important to
understand the
context of the
individual's
presentation.
For this reason,
FDA encourages
you, the open
public hearing
speaker, at the
beginning of
your written or
oral comment to
advise the
Committee of any
financial
relationship
that you may
have with any
company or group
that may be
affected by the
topic of this
meeting.
For example,
this financial
information may
include a
company's or a
group's payment
of your travel,
lodging or other
expenses in
connection with
your attendance
at this meeting.
Likewise, the
FDA encourages
you at the
beginning of
your statement
to advise the
Committee if you
do not have any
such financial
relationship.
If you choose
not to address
this issue of
financial
relationships at
the beginning of
your
presentation, it
will not
preclude you
from speaking."
At this time I
believe we have
two speakers. We
have a Mr. Hooks
and a Mr.
Weidman, is that
correct? Please
come forward and
introduce
yourself. At
this time
indicate your
affiliation.
Each speaker is
allotted a 15
minute period.
MR. HOOKS: Good
morning.
I don't have any
financial things
with anybody,
nobody paid for
my way.
What we'd like
to do is address
the military
application for
aspects of the
jet gun
injectors.
I represent
HCVets. com.
It's a website.
Go to the next
one. All right.
I'm getting
ahead of myself.
Anyway, what we
do is we have a
website that
allows veterans,
military
members, their
families or
whatever to seek
information on
the
contamination or
infection of
hepatitis C via
the jet guns.
If you look at
this chart here
you'll see that
the majority of
the people that
have hepatitis C
are veterans,
the largest
portion of
Vietnam era. The
reason that
occurred is if
you think about
the military at
the time, was
probably at
their peak. The
one thing we all
share in common
is we were all
inoculated with
the jet guns.
The other thing
is when you look
at most of the
studies
referring to
this stuff
you'll see they
mention
hepatitis B and
HIV. Well,
hepatitis C is
more infectious
than HIV, it's
also a lot
harder. It's a
lot harder to
get rid. So the
cleaning and all
like that is
very important.
Next one,
please.
If you look at
the VA
Administration
and all like
that statistics,
there's 25
million plus
veterans still
alive in this
country. Only
about ten
percent of these
folks go to the
VA. So the
numbers that
you're going to
see are smaller,
I believe,
because there
are a lot of
veterans who do
not use the
Veterans
Administration
as their health
service.
If you look at
the numbers from
the CDC and the
Veterans
Administration
right now, out
of the 25 or so,
comes out to
about 458,000
veterans have
hepatitis C. But
I think the
numbers would be
quite larger
than that if you
took into
account the
whole
population.
Next one,
please.
If you take the
numbers in with
all, add the 2
percent and
everything like
that for the
population, of
course there's
really been no
studies on this
since '94, so
these numbers
wee taken with
the CDC at
different times
and all, and the
2 percent was
added, you know,
for the
population
growth and stuff
like that. And
you come up with
these numbers
here. Out of the
33.4 million
veterans or
people that are
infected with
hepatitis C, 2?
million would be
veterans.
Next one,
please.
Based on the
infection rates
quoted by the VA
and the CDC,
approximately 75
percent of the
estimated people
with hepatitis C
are military
veterans with
infections
longer than 20
years. Out of
the estimated 3
million
chronically
infected stated
by the National
Institutes of
Health, an
estimated 2.2
million had this
disease for over
20 years, a
projected 20
percent or
450,000 veterans
are expected to
develop
sclerosis or
90,000 are
expected to
develop cancer
now.
Next one,
please.
I'm sorry about
the picture. It
didn't come up.
It was a graph.
The role of the
jet gun in the
transmission of
hepatitis C. The
Ped-O-Jet was
introduced about
1950s, developed
under a U.S.
military
contract for
mass
vaccinations of
recruits of 600
to 1,000
injections per
hour. The WHO
document says an
hour and a half.
If you go on an
hourly basis,
that's about six
injections at
600 or 3.6
injections a
second per hour.
If you go to an
hour and a half,
it's 9 seconds
per injection or
5.4 injections
per second.
That's a
relatively rapid
fire. I think
anybody's that's
been around in
those lines
understand
there's no time
to waste. Real
close quarters
and you're
hustled through.
Next one,
please.
This is a
picture of the
old apparatus
that was used. I
believe up until
about '94. It
wasn't me.
Okay. Next one,
please.
The Air Force
Infectious
Disease and
Control
Epidemiology
Board,
Department of
Defense Wide
Review of
Vaccine Policies
and Procedures
said that
injector nozzles
were frequently
contaminated
with blood. What
they did is they
had -- I think
it was probably
a surprise visit
to Parris
Island. And they
witnessed a mass
injection of a
lot of recruits
coming in. And
they noted in
that document
that there the
nozzles were
frequently
contaminated
with blood.
There were no
wiping or
precautions
taken.
Next one,
please.
The problem with
the jet injector
gun during the
Board meeting in
1986, Captain
Michael Stek,
Jr., MC, USN
presented data
and press
clippings to
suggest that
contamination of
the jet injector
gun which had
been used in a
private clinic
in California in
1985 was
responsible for
causing
hepatitis in 64
patients. The
possibility was
also raised that
HIV infection
might be
transmitted by
the jet gun when
biological
products such as
gamma globulin
were
administered. In
numerous
meetings the
board
recommended in
1988 that an
injector gun be
used only by
authorized
military and
technical parts
and sterilized
according to
standard
procedures.
Next one,
please.
What are the
standard
procedures for
the jet
injections?
Next one,
please.
That would the
manufacturer's
recommendations.
Next.
The
manufacturer's
recommendations
recommended the
devices be wiped
in between each
injection. There
was a meeting, I
guess, of this
organization in
'99 where a
representative
of the company
was here and
they stated that
in 35 years they
were always
wiped and never
had an issue.
I'd like to
bring out at
this point in
time probably
you never had an
issue with
hepatitis C by
the simple fact
a majority of
people are
asymptomatic and
it takes decades
before you find
out you've got a
problem.
Thirty-five
years is not a
stretch in this
area. The
majority of the
people won't
have a problem
until at this
point in time.
There was a
study done in
England where it
came out that
they could
infect 31 out of
a 100 if the
guns weren't
wiped. There was
a statement made
that there's
nowhere in the
world recorded
that the guns
weren't wiped.
Well, we have --
the next one,
please.
The website did
a survey, and
this a partial
selection of
people that
answered the
survey. We have
answers from
medics that
administered the
shots and
received the
shots, we have
all different
bases and
military
branches, and
comments from
the individuals
that state the
guns were not
wiped. I
personally can
attest to that.
They didn't wipe
them before they
nailed me or
anybody before
or after me.
Next one,
please.
The expectations
fell short. As I
stated earlier
the people in
charge of the
basis and the
medical, and
stuff like that,
were under the
idea that the
guns were being
wiped in between
each injection.
That's not the
case. The human
error factor,
for whatever
reason, the
things weren't
followed. I've
talked to some
medics that had
this duty when
they were in the
military, and
this is what
they considered
to be a great
job. You go in
in the morning,
you throw a
bunch of shots
out, you get
done early. You
got the rest of
the day off. You
know, that was
just the way
they looked at
it. There was no
harm, no fault
in my mind
because they had
no idea with the
little bit of
training they
had what they
were doing. They
had no
understanding of
the infection
rates. Hepatitis
C at the time
wasn't even
something
described. You
were non-A,
non-B if you
were diagnosed
at all.
The next one,
please.
In dealing with
the VA, it's
been an uphill
battle for a lot
of folks because
the simple fact
is they don't
fit into the
prescribed
methods of
transmission for
hepatitis C. The
CDC and all have
kind of left out
a whole
generation of
folks, and it
makes extremely
hard for someone
who has no other
reason except
for their
injections, to
get hepatitis C.
Back in 2003
there was a
claim that was
based solely on
the jet
injectors. The
veteran won that
one, but it had
to go to
Cleveland to the
Tiger Team to
get there.
Next one,
please.
Here's some of
the
documentation
that was used
and the studies
that were used
to validate the
claim.
I'd like to
mention, too,
besides the hard
copies, I have
CDs that if you
go on line the
links will work
and link you to
these studies.
It would take
too long to get
into them.
Next one,
please.
This is the
DoD's
needle-free
injection policy
chronologically.
It shows when
they started to
stop using the
jet injectors
and the reasons
why. The dates
and the
organizations,
and their orders
that came out.
Once again, you
know, the links
will take you to
the full study.
Next one,
please.
Okay. For
infection rates
we're talking
picoliters of
blood, that's
very small. It
doesn't take a
lot. And there's
been numerous
studies on that.
Hepatitis B,
basically, can
be transmitted
at about 10
picoliters.
Hepatitis C runs
in, I believe,
at about 35 or
HIV at about 40.
Somewhere in
that range.
There hasn't
really been any
hard studies
that I've seen,
or found or
heard about that
relates to
hepatitis C.
That's something
that really,
really needs to
be looked at
because it's not
a problem that's
going way. I
mean, this whole
thing with me
not knowing that
I was infected,
I in turn
infected my
wife. She wasn't
real happy about
that, but I'm
not the only one
that has done
that not
knowing. I've
donated blood up
until like '92,
and then I
stopped for
physical reasons
not because I
was tested with
hepatitis C. So
we have a larger
epidemic then
what's showing
up in the
numbers. And it
really needs to
be looked at. We
have to stop it
any way we can.
And by ensuring
that these guns
or any other
device that has
the ability to
transfer blood
in any amount is
designed in a
fashion that
can't happen. I
don't want
anybody else to
have to go
through what
I've been
through, or a
bunch of other
fellows, either.
Next one,
please.
This is a CIA
report, which
once again the
link will take
you to. What we
have here,
basically they
did a study in
the areas of the
sub-Sierra and
Southeast Asia,
and stuff like
that. They had
an upheaval with
HIV, and all
like that.
The other
problem you'll
see and where
our folks are
right now
serving us with
great courage,
they're also hot
beds for
hepatitis C. I
think that the
fellas and gals
that are over
there now should
be checked. If
they've had any
injections and
stuff, they
should also be
checked and nip
in the bud
before it gets
like it did with
us 20/30 years
down the road.
Next one,
please.
Once again, this
study is taken
not in this
country, we
really haven't
taken the time
to do in depth
studies for
hepatitis C. We
have some on HIV
and some on
hepatitis B. So
most of the
studies you'll
see are from
foreign lands.
We haven't
really addressed
it
appropriately.
Next slide,
please.
That's my idea
of the beautiful
world and all
reality. Like I
said, any device
that transfers
blood, the
needle jets
specifically,
they need to be
addressed
appropriately. I
know there are
some
modifications
that have been
made like caps
and disposable
ends and stuff
like that. I've
seen where
they're working
on things. But
they really do
need to make
sure these
things don't
transmit blood
in any fashion.
That's all I
have.
CHAIRMAN
EDMISTON: Thank
you very much,
Mr. Hooks.
MR. HOOKS: Thank
you.
CHAIRMAN
EDMISTON: At
this time I'd
like to invite
members of the
Panel who may
have questions
or
clarifications
of Mr. Hooks'
presentation to
please address
the speaker. Are
there any
questions from
members of the
Panel?
Thank you very
much.
Do we have any
other speakers
who wish to
address the
meeting?
I think at this
time since we're
ahead of the
game here, we're
going to go
ahead and take a
brief 15 minute
break. The next
presentations
will be from the
FDA, and there's
a continuity of
those
presentations so
I'd rather not
break them up.
So let's take a
15 minute break
and convene at
9:15.
(Whereupon, at
9:00 a.m. a
recess until
9:17 a.m.)
CHAIRMAN
EDMISTON: I
think we'll
reconvene the
meeting now. I'd
like to ask all
the Panel
members to take
their seats,
please.
I'd like to make
a very brief
announcement. It
was initially
announced that
Dr. Weniger from
the Centers of
Disease Control
would be here
giving a
presentation.
But,
unfortunately,
he will not be
able to be here
to make that
presentation.
We will now
proceed to the
FDA
presentations
for the Panel.
The first
speaker will be
Mr. Anthony
Watson, Chief of
the General
Hospital and
Personal Use
Devices Panel.
Mr. Watson?
MR. WATSON:
Thank you. And
I'm just going
to introduce the
speakers. We
have three
speakers today.
Mr. Jason Lipman
is an engineer
in the General
Hospital Devices
branch. He will
be discussing
the regulatory
history of jet
injectors.
Then we have Dr.
Shewit Bezabeh,
who is a medical
officer in our
division. And he
will discuss the
safety history
with these
devices.
And then
following him
will be Dr. Daya
Ranamukha, who
is a
microbiologist.
Is that correct?
Molecular
biologist. I
apologize. A
molecular
biologist from
our Office of
Science and
Engineering
Laboratories.
And he will
discuss
potential
methods for
testing for
these devices.
So now I'd like
to ask Mr. Jason
Lipman to come
to the podium,
please.
MR. LIPMAN: Good
morning. My name
is Jason Lipman.
I'm reviewer in
the General
Hospital Devices
Branch. If you
haven't figured
it out yet,
we're here to
talk about jet
injectors.
CHAIRMAN
EDMISTON: Excuse
me. Could I ask
you to speak
directly into
the microphone.
MR. LIPMAN: Oh,
sorry.
CHAIRMAN
EDMISTON: We're
having some
problem hearing
you.
MR. LIPMAN: Is
that better?
CHAIRMAN
EDMISTON: Yes.
That's great.
MR. LIPMAN:
Okay. Jet
injectors are
also known as
needle-free or
needleless
injectors. As
defined by the
Code of Federal
Regulations a
jet injector is
a
nonelectrically
powered device
used by a health
care provider to
give a
hypodermic
injection by
means of a
narrow, high
velocity jet of
fluid which can
penetrate the
surface of the
skin and deliver
fluid to the
body.
Next, please.
Jet injectors
are Class II
devices. They
regulated
through the
5.10(k)
premarket
notification
process. And jet
injectors must
demonstrate
substantial
equivalence.
Next, please.
There are two
main types of
jet injectors.
There are single
use devices and
there are
multiple use
devices.
Single use
devices are
devices in which
the entire
device is
discarded after
one use.
There are three
types of
multiple use
devices. There's
single use
cartridge
devices in which
the fluid
contacting
components are
discarded after
one use. There
are devices that
are labeled and
sold for only
one patient.
These devices
can be multiple
use, but only
one patient is
using them. And
there are
devices that
have a reusable
fluid path. As
indicated by the
yellow, these
are the devices
that we will be
focusing on
today. These
devices
typically have a
large medicinal
vial that fills
an injection
chamber after
each subsequent
injection.
Reusable fluid
path injectors
are also known
as multi-Use
Nozzle Jet
injectors or
MUNJIs, for
short.
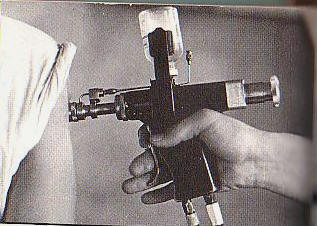
Here's a picture
of a bunch of
jet injectors.
As you can see,
many of them do
have that
medicinal vial
at the top of
the injector
which I just
mentioned.
Next, please.
I want to talk a
little bit about
how a jet
injector works.
Jet injectors
must create high
pressure,
usually by the
use of springs
or compressive
gas. This high
pressure forces
the medicinal
product out of
an injection
chamber through
an orifice and
into the body.
There are four
target tissues
for injectors;
mucosal
membranes,
dermal tissue,
subcutaneous
tissue,
intramuscular
tissue.
Next.
There are two
primary uses for
MUNJIs. That's
immunization and
administering
anesthesia
during dental
procedures.
There are
several
advantages of
MUNJIs use. They
include high
delivery rates.
It doesn't take
very long to
prepare for a
subsequent
injection.
There are
several
needle-free
benefits for
MUNJIs use.
There's no reuse
of needles, no
chance of
contaminating
needle-stick
injuries. And
there's no
patient fear of
needles because
there,
obviously, is no
needle.
There's a
reduction of
volume of
clinical waste.
And these
devices are
economical
because the
device is
reused.
There are a
couple of
disadvantages
for MUNJIs. The
focus of our
presentation
today is the
first one, the
potential for
blood
cross-contamination
or disease
transmission.
The second is
the potential
for laceration
injury from
improper
technique. And
this can occur
since the jet
stream has such
a high velocity
of jet stream
that if you were
to actually lift
it off the skin
prematurely, you
could lacerate
the skin from
that high
velocity jet.
Next, please.
There has been
one documented
case of
cross-contamination.
This was in
California in
1985 at a weight
loss clinic. It
resulted in a
hepatitis B
outbreak. In
addition to that
outbreak, there
have been in
vivo animal
studies and
bench laboratory
studies that
also link these
devices to
disease
transmission.
This will be
talked about in
more detail by
subsequent
presenters.
So I want to
talk about how
the
cross-contamination
occur. It can
occur, as we
heard before,
about blood
actually the
skin contacting
surface on the
injector or that
blood or serum
can actually go
up into the
fluid path. And
there a couple
of theories as
to how that can
actually occur.
One is
splash-back.
Again, the high
velocity jet can
actually bounce
back off the
body and back
through the
small orifice.
Or there's also
a thought that
the injection,
the pocket of
fluid in the
body is
pressurized and
pressurizes the
tissues around
it and those
tissues can
actually push on
the fluid and
push back up
through the
orifice.
In either way,
the residual
infected blood
or serum can be
injected into
the subsequent
patient causing
a blood-born
illness.
Manufacturers
have attempted
to mitigate that
risk of
cross-contamination.
The primary
design of the
mitigations are
single-use
patient
contacting
components, such
as caps, spacers
or sheaths. But
there have been
no validated
methods to
assess the
effectiveness of
these
components.
Next, please.
So the challenge
of evaluating
the potential
for disease
transmission
exists because
there's no
consensus on the
amount of blood
contamination
that can
potentially
transmit
disease, and
there's no
validated test
method for
detecting blood
cross-contamination.
And, again, this
will be talked
about in more
detail in
subsequent
presentations.
There is global
concern about
using these
devices, the new
devices as well,
the new MUNJIs.
The World Health
Organization
recommends
against MUNJIs
use. The Centers
for Disease
Control and
Prevention
recommend
weighing the
risks versus the
benefits; the
risks of typical
syringes and
needles versus
the jet
injectors.
Hopefully, I'm
hoping that Dr.
Martin Friede
will talk in a
little bit more
detail about
their current
policies.
In 1999 the FDA
held an Advisory
Panel meeting to
discuss the
guidance for jet
injectors. This
was talked about
earlier today.
This was to
figure out the
evaluation
criteria that
would be
documented in
our guidance
document for
evaluating jet
injectors.
During this
Panel
presentation, or
this Panel
meeting we also
discussed the
potential for
cross-contamination,
what we're here
to talk about
today.
At the end of
that meeting
there were two
recommendations
made by the
Advisory Panel
to the FDA
relating to this
issue. The first
was to consider
the postmarket
surveillance. We
have reviewed
all of the
medical device
reports on this
issue. There has
only been one
medical device
report related
to
cross-contamination,
and this was
actually a case
of misuse and
did not result
in any
blood-born
disease, at
least documented
blood-born
disease.
Could you go
back for a
second? Thank
you.
The second
recommendation
to the FDA was
to investigate
the possibility
of developing a
standardized
methodology to
determine
contamination.
We have reviewed
all the current
methods and even
looked at some
future methods,
and we will be
talking about
this more later
today. But to
date there are
no validates
test
methodologies
available.
Next, please.
These are the
references that
I've cited in
this
presentation.
Next, please.
I just want to
talk a little
bit about the
purpose of
today's meeting.
We're here to
discuss the
cross-contamination
risk associated
with MUNJIs and
to discuss the
methods that
might be used to
assess this
risk.
This concludes
my presentation.
I hope it gave
you a good
background for
what we're going
to discuss
today.
At this time I'd
like to call up
Dr. Shewit
Bezabeh who will
give a clinical
perspective on
this issue.
Thank you.
DR. BEZABEH:
Good morning. My
name is Shewit
Bezabeh. I'm a
Medical Officer
with CHRH, the
FDA.
My background is
both public
health and
epidemiologist.
Also I'm an
internist. I'm
also active in a
clinical
practice. I have
been with the
FDA for the past
four years as a
Medical Officer.
Next slide,
please.
Today I will
give you an
overview of
MUNJIs. The
device has
history. The
public has need.
The
effectiveness
experience with
these devices,
the history of
safety concerns
and the concerns
for current use.
Next slide,
please.
Jet injectors
are needle-free
delivery devices
that facilitate
the
administration
of medications
under high
pressure stream
into tissue.
These devices
can administer
vaccines and
other
medications into
subcutaneous
tissue,
intramuscular
tissue and also
dermal tissue.
People have
categorized
these devices
into three
categories. The
first one, the
first one I'll
use is usually
used for single
use can also be
reused with the
same person. We
see these
devices being
used with a
number of
diabetics.
The second
category is low
work load. About
30 injections
per health care
worker.
And the third
category, which
is the focus of
today's meeting,
will be high
work load,
injection of a
100 injections
per health care
worker.
Next slide,
please.
The history of
these devices,
they start in
the 1860s, was
initially
developed in
France to
administer a
number of
liquids.
In 1936 the
first jet
injection device
was attempted in
New Jersey.
In the '40s the
first
commercially
available jet
injector was
Hypospray. It
was initially
devoted for
single use, self
administration
for diabetics.
It was designed
to overcome
childhood needle
phobias.
From the
mid-'40s to the
'60s it was
introduced
massively into
the military for
clinical use.
From 1976 to
present up to
now it is
cleared by the
FDA as a Class
II pre-amendment
medical device.
The need for
public health
for these
devices is
multiple -- for
a number of
reasons. These
devices are
needle-free, so
they avoid the
needle entrance
risk due to
needle injuries.
Globally there's
high risk with
needles and
syringes because
of improper
recycling, and
also reuse with
proper
sterilization.
WHO experience
with that, half
of the
injections in
the developing
world are unsafe
and result in
about from 8 to
16 million
hepatitis B
virus infections
per year, 2.3 to
4.7 million
hepatitis C
infections and
about 100,000
HIV infections.
In the U.S.
there are about
87 health care
workers contract
hepatitis B
virus due to
occupational
exposure, of
this there's
about 200 cases
per year.
The risk of
infection after
a needle stick
injury with an
infected blood
for HIV is about
3 in a 1,000,
hepatitis C the
range was from 1
to 7 percent and
hepatitis B,
which is the
most highly
infectious,
about 30
percent.
The other aspect
of need for
these devices,
they can be used
in response to
bioterrorism
because they can
rapidly immunize
first
responders,
exposed
populations.
They can be used
in pandemics,
regional
epidemics and
emerging
infections. They
have been used
with
meningococcal
meningitis,
yellow fever,
influenza.
There is a
global need for
this
eradication.
They have been
used with polio
initially. Polio
is almost
eradicated.
Measles is
targeted for
eradication.
Many of the
program for
immunization
vaccines
practices
require
injections. And
also as
mentioned
earlier, needles
and syringes
have a number of
limitations.
They also have
potential need
for future newer
vaccines. They
have been tested
for malaria DNA
vaccines and
also for
emerging
vaccines such as
when the vaccine
is available for
SARS and other
infections.
The advantage of
this device is
included, it has
a potential high
rate of
vaccination.
They can
vaccinate over
600 people per
hour. Can
respond to
pandemics,
regional and
local epidemics.
Can also respond
rapidly to
bioterrorist
attack. Can
administer
off-the-shelf
vaccines.
They have a long
history of use
with many types
of vaccines.
They can be
filled at the
end user or by a
manufacturer.
They eliminate
the needle stick
risk and sharp
disposal burden.
They are also
very cost
effective
compared to
needles and
syringes.
The main
disadvantage,
which is the
focus of today's
meeting, is the
potential for
blood
cross-contamination.
Also, they're
believed to have
increased pain,
especially the
adjuvant added
vaccines as
compared to
needles to
syringes. Also,
improper
technique may
result in
laceration of
injury. They're
believed also
more reactogenic
than needles and
syringes. You've
seen increased
erythema
hematoma
bleeding at the
injection site.
Immediately you
see more
erythema and
hematoma. Some
of the delayed
reactions
includes
soreness,
erythema in
duration and
edema. Other
local adverse
events include
bleeding of
injection site.
As mentioned
earlier, there
could be
laceration,
especially if
improper
technique is
used. And there
have been very
rare reports of
traumatic
injuries.
In terms of
effectiveness
expense. We have
over 50 years of
device use
delivering
millions of
injections.
There have been
a number of
studies which
demonstrate
effectiveness of
these devices,
mainly by
measuring immune
response and
immunogenicity.
We have a number
of randomized
control trials,
review of
clinical trials.
Also respected
comparative
studies.
I should note
that even though
these studies
assess the
effectiveness of
these devices,
none of them
have studied for
their
safety.
In terms of past
use, the U.S.
Department of
Defense from
1965 to 1980
have given about
20 to 40 million
injections to
military
personnel. The
global smallpox
eradication
program, 50 to
100 million.
During 1976
swine flu
epidemic, about
75 million have
received
vaccination
using the
device. The
African
Meningitis
Program, 1988
through 1998,
about 80
million. The
Brazilian
Measles
Eradication
Program, an
estimated 60 to
80 million
people have
received
vaccination with
this device. And
globally, from
100 to 500
million have
used this device
to receive
vaccinations in
the past 30
years.
Even though we
have extensive
history of use
and effectively,
there have never
been no
surveillance
implemented to
assess
transmission
potential
between this
use.
Next slide.
Some of the
vaccines that
have been used
with this
device, include
both light and
inactivated
vaccines,
measles, mumps,
rubella, yellow
fever. Some of
the inactivated
vaccines include
botulism,
cholera,
hepatitis A and
B, influenza and
others.
Next slide.
In terms of the
history of
safety concerns,
in the late '60s
early '70s
people started
noticing blood
on the nozzle of
these devices
which was
initial concern
for the purpose.
There was only
one documented
disease
transmission
which occurred
in California in
1985. A cohort
of patients were
receiving formal
injections, had
clearly
documented
hepatitis B
virus. We
believe this
transmission was
through this
device.
In addition, we
have some
experimental
evidence as well
as some
epidemiologic
evidence
implicating this
device, this is
transmission.
Some of the
experimental
evidence include
in 1985 Brink
coworkers took
mice which were
clinically
infected with
LDH virus. They
had a cohort of
mice received
injection and
they were able
to demonstrate
that 16 out of
49 mice had
acquired LDH
virus through
that injection.
In 1988 Zachoval,
which have
reported in the
Lancet, took 5
patients. Four
of them had
positive
serologic
markers for
hepatitis B. The
fifth patient
was HIV
positive. They
injected them
with a jet
injection device
and then they
tested the
nozzle and the
injection site.
While the nozzle
was negative for
any markers,
three out of
four of the
hepatitis B
carriers, the
injection site
was positive for
hepatitis
markers and the
HIV patient also
positive for the
marker. The
theory being
that if there
had been a
subsequent
injection, these
markers would
have been
transmitted to
the next
patient.
Next slide,
please.
Some of the
epidemical
evidence so far
include the 1994
about 2800
subjects were
receiving
routine
immunization via
jet injector.
The injected was
tested instead
of giving it to
the next
subject, it was
tested. It was
collected in a
test tube and
tested for
blood. And about
28 of them,
which is about
one percent of
the subject
recipients
tested for
occult blood.
In 2001 there
was an
epidemiological
survey done in
Brazil where
about 750
patients where
hepatitis B
virus carriers
had a
multi-variant
analysis to
evaluate the
risk factor for
transmission.
And out of the
multi-variate
analysis, a
cohort of people
who had received
prior yellow
fever
vaccination via
the jet injector
was a risk
factor as for
hepatitis B
infection.
Again,
implicating the
device as a
vector for
disease
transmission.
A field study
done in Brazil
again to look at
the safety of
the injector.
This
investigator
took two modes
of injection
type. One they
took
noncompliant
stimulating no
interference
between the
device -- I
mean, vaccine
delivery. And
the second mode
was a confirmed
compliance mode
where the nozzle
of the device
was swabbed with
alcohol.
And they took
the volunteers
and injected
them with
buffered saline
and they
collected three
subsequent
injected into a
test tube and
tested the
ejected for
blood.
In the first
injection in the
noncompliant
mode, about 30
out of 117
patients, which
is about 11
percent, were
positive for
occult blood.
And then in the
compliant mode,
9 out of 117
patient, which
was about 8
percent, was
positive for
blood.
In the second
injected, about
4 percent, 4 out
of 117 in the
noncompliant
mode. And the
second mode, the
complaint mode,
3 out of 117,
which is 2.5
percent were
positive for
blood.
Whereas, the
third injected
there was no
blood positivity.
Again, even with
interference
alcohol swab, as
you can see both
the first and
second injected
in both the
noncompliant and
compliant mode
there was blood
positivity.
Again,
implicating that
this device
possibly
delivers
transmission.
In 1999 the
Armed Forces
Epidemiology
Board observed
frequent blood
contamination of
the nozzle in
high volume
recruit
immunization.
Next slide.
Based on this
and other safety
concerns and
other studies, a
number of
agencies come up
with policies.
In 1987 WHO
restricted
device use. In
1996 WHO also
stated that
MUNJIs is not
recommended for
mass use. In
1997 the U.S.
military
withdrew the use
of the device.
In 1999 FDA had
a Panel
presentation
meeting, as
mentioned
earlier. And the
Panel meeting
was to discuss a
guidance
document.
However, also
the safety of
this device were
discussed. And
the Panel had
two
recommendations.
The first
recommendation
was to continue
to do postmarket
device
surveillance.
And the second
recommendation
was to
investigate the
possibility of
developing a
standardized
methodology for
the safety of
the devices.
In 2002 the CDC
Advisory
Committee for
Immunization
Policies
discussed the
use of these
devices and they
stated that
MUNJIs use
should be
limited weighing
the risk versus
benefit of
MUNJIs with
needles and
syringes.
And most
recently, 2004,
WHO had also
discussed the
use of these
devices. And the
conclusion was
it would not be
possible to
adequately
endorse the
safety of these
devices.
Next slide.
In terms of
blood-born
transmission, we
know that
hepatitis B
virus is more
infectious than
HIV and
hepatitis C. And
we also know
that there about
9 to 10 to 11
hepatitis B
virus DNA copies
per cc, which is
about 1 to 100
hepatitis B
particles per
picoliter. There
have been
studies where
they have
estimated that
10 picoliters
being the
smallest amount
for transmission
of hepatitis B
virus.
Next slide.
A number of
assays have been
tested and tried
in the past to
measure the
blood injected
as a marker for
assay. Some of
the assays
include serum
albumin
measurement as
an indicator of
blood. There
have been
sensitive ELISA
assays. However,
there is no
acceptable limit
of blood
detection to
demonstrate
safety of these
devices. Some of
the proposals
which have put
forward include
to do clinical
trials with
hepatitis B
positive
population and
also to test
injected for
hepatitis B
virus by PCR.
However, we're
not sure if
these assay
models are
sufficient to
evaluate the
safety of these
devices, also
are there any
testing methods
to assay the
safety of the
devices.
The next
presenter, Dr.
Daya Ranamukha
will go further
into the testing
methodology and
assays.
Thank you very
much.
DR. RANAMUKHA:
Thank you.
The title of my
talk today is
potential safety
evaluation
strategies for
MUNJI devices.
I'm Daya
Ranamukha-arachchi.
I'm a molecular
biologist at the
Office of
Science at the
Center for
Devices and
Radiological
Health. I have
over ten years
of experience in
molecular
methods in human
genomics.
So going back to
the percentage,
and Dr. Bezabeh
before and
others talked
about safety
concerns for
MUNJI use, and I
want to stress
the important
points here
again.
MUNJI can exert
local adverse
events and it
could be delayed
for early
reactions, and
again can lead
to bleeding at
the injection
site. These are
more common in
MUNJI devices
than
needle/syringe
devices.
What I'm going
to talk to you
today about
mainly, the risk
of
cross-contamination
with blood. So
I'm going to put
forward the
potential
evaluation
strategies in
this context.
Next, please.
So the first
question that
comes to your
mind is when you
think about
cross-contamination,
is there safe
limits of blood
cross-contamination?
Insights into
this comes from
virology data.
If you look at
the hepatitis B
carriers, they
contain around
10 to the 9 to
10 to the 11 DNA
copies per
milliliter. If
you go down in
the volume, it's
about 1 to 100
HBV copies per
picoliter of
blood. But it
can also go in
some carriers,
they go to like
10 to the 15 DNA
copies per
milliliter.
And there's one
study that shows
HBV may be
transmitted with
as little as 10
picoliter of
blood and using
one animal
model. This was
published in
1984.
So when you
combine these
two facts, is
the 10 picoliter
of blood or 10
to 1,000 HBV
copies the limit
that we want to
dictate? And
then the next
question is are
there test
methods to
achieve the
required limit
of detection?
These are the
questions that
we need to
address.
Next, please.
So if one were
to evaluate the
contamination
risk, what are
the challenges
we have to face
between use
cross-contamination.
So these are
whole list of
questions that
comes to one's
mind; collection
of sample; how
we can collect
the samples to
evaluate between
use
cross-contamination.
Then what are
the analytes?
What are the
molecular
methods? Then
when you think
about the
molecular
methods, what
are the limits
of direction and
accuracy,
specificity and
reproducability?
And then finally
depending on all
these answered,
what are the
safe acceptance
limits? Is there
acceptance
limit?
So addressing
all these
issues, I have
divided my talk
into three
categories.
First, analyze
for testing.
Once we collect
the samples, how
we can look at,
what are the
analytes that we
need to test? Of
course, blood
markers and then
we can think
about pathogenic
contaminates,
what are the
markers? Then
what other
methodologies;,
molecular
methods
available to
determine
contamination?
Serology-based,
then DNA
amplification
based and
combined
approaches
including DNA
hybridization
technologies.
Then the third,
cross-contamination
study designs.
We have animal
models, we have
human models. So
we can look at
all that.
So coming back
to the first
part of my talk,
analytes.
Obviously, many
talk about blood
cross-contamination
comes to our
mind the blood
markers. So we
can look at
blood markers
like abundantly
available
proteins such as
serum albumin as
surrogate marker
for the presence
of blood. So
this has been
done before
actually using
sensitive ELISA
methods. But the
disadvantages of
using this serum
albumin is it
can create false
positives, false
negatives and
deduction
limits. False
positives in the
sense that serum
albumin presents
in everywhere
saliva and skin
cells, so this
can create false
positives.
Then the false
negatives. Under
cold storage
conditions serum
albumin can bind
to collection
tubes. So then
when you think
about ELISA, the
detection limits
if very narrow.
So you have to
go through a
series of
dilutions in
order to get
within the
dynamic range of
detection.
Then with regard
to blood
molecules, I
want to stress
the point that
what is the
limit of
detection 10
picoliter of
blood. This
number came from
one single study
using one
chimpanzee. This
study was not
meant for
actually looking
at HBV
transmission,
but to evaluate
it was
methodologic
paper looking at
ELISA versus DNA
detections. And
in that what
they did was
they the serial
disillusions of
the saline,
buffered saline
and they found
out that 10 to
the minus 10
dilution they
could infect one
chimpanzee. But
their aim, the
object of the
study was to
evaluate how
good at
analyzing
detecting HBV
contamination.
So what I mean
to say here is
that this 10
picoliter blood
limit is not
statistically
validated.
So coming to the
next one, then
the second class
of analytes is
the viral
markers which
has the highest
contamination
potential. For
this, the
infections come
from needle
stick injuries.
If you look at
HIV HCV, HBV,
HBV has the
highest
potential with 6
to 30 percent
depending on the
status of the
contaminating
blood.
Then HBV has the
highest
potential for
transmission due
to
cross-contamination.
Which is the
most prevalent?
Over 2 billion
people are
infected with
more 350 million
chronic
infections based
on the WHO
report.
Survivability is
high and can be
easily
integrated into
the host genome.
Next, please.
So last year's
WHO injector
safety meeting
they come to
consensus that
HBV is an
appropriate
marker for
determination of
injector safety.
There are also
certain group of
advantages. If
you are using
HBV as an
analyte, there's
presence of
international,
WHO
international
standard and
then
availability of
quality control
panels. And
availability of
molecular assays
for HBV
detection.
There are
internal
controls, such
as murine
cytomegalovirus
for evaluation
of false
positives as
well as false
negatives.
So all these
advantages lead
us to develop
good test
methods if you
need to.
Next, please.
Now the second
part of my talk
is the test
methods. What
are the test
methods
available? There
are a whole host
of methods
available based
on serology, DNA
amplification
and combined
approaches using
DNA
hybridization.
Now what I have
summarized here
in this table is
the more
sensitive
methods with the
principle --
actually those
principle
technologies.
Serology based
uses serum
antigen, surface
antigen and also
e antigen.
So then other
methods uses HBV
DNA. So the
samples either
serum or plasma.
Then the limits
of the
detection, I
want you to look
at the last two;
real time PCR
and NAT
technology. NAT
technology is
the nucleic acid
testing
technology based
on PCR, DNA
amplification,
plus DNA
hybridization.
So these limits
of detection, I
got it from
published data
which gives like
100 copies to 10
to the 7 and 10
to the 9
sensitivity
limits of
detection.
So I want to
stress the point
here that
there's test
methods
available for
single copy
detection. If
you want to look
at single copy
detection, there
is no test
methods
available.
Now next slide,
please.
I just wanted to
put this slide
because what
other emerging
technologies can
do in this
context.
Obviously,
nanotechnology
comes into play.
And there's some
published
studies, one for
DNA detection
called
biobarcode DNA
detection which
can detect DNA
at 500
zeptomolar
level, which is
a quality
detecting all
available copies
in a solution.
Then again, when
you look at
protein
detection using
the same
technology, you
can detect
antigens at
atomolar levels.
So these are
only research
tools which is
published
recently. These
have not been
validated under
any diagnostic
setting.
So the next
slide, please.
And I want to
stress this
point before I
move on to the
next category.
Molecular
methods for HBV
testing.
Molecular
methods have a
lower limit of
detection than
conventional
assays. And
MUNJI
cross-contamination
may be
investigated
using HBV-NAT or
Taqman assays.
However, this
has yet to be
validated. Then
there are
studies to
establish
performance
characteristics
of these assays
for HBV
detection in
MUNJI device use
have not been
conducted.
Next please.
Now I want to
switch the gears
here to talk a
little bit about
what are the
possible study
designs we can
look at. This is
the last part of
my talk.
So we can look
at animal versus
clinical
studies. If you
look at animal
studies, what
advantages does
it give? Provide
well controlled
biological
uniform study
designs and we
can directly
evaluate viral
transmission
potential.
However, we are
to take into
consideration
the substantial
histologic
differences that
exist between
human and
animals schemes
and muscle
development.
So then the
clinical
studies, on the
other hand, use
the direct
impact of
injected device
on
cross-contamination
in humans.
Genetic
variables are
also taken into
consideration.
But it is unable
to get IRB
approval for
direct human
evaluation of
viral
transmission.
So next slide,
please.
There's one
published model,
actually, using
animals for
evaluating
cross-contamination
potential of
MUNJIs devices
in this study
which was
published in
2001 in Vaccine
by Hoffman et
al. They used
cows, young cows
of 8 to 12 weeks
and they used
the same set of
cows repeatedly.
And what they
did was instead
of using the
vaccine, they
used a phosphate
buffered saline
in a buffer at
.5 milliliter
per dose. And
then t hey
injected to one
calf and then
collect the next
ejectate before
injecting to
another one into
a separate
container.
That's in the
real world
situation in an
immunization
program, that's
the one that
goes to the next
person. So they
collected that
and then
evaluate the
blood markers,
surrogate
markers, serum
albumin by
sensitive
analysis. And
then they
compared the
results with
negatives based
on preinjection
doses.
Next slide,
please.
So using this
same method we
can think about
potential
clinical studies
for evaluating
cross-contamination.
There are two
types we can
look at. If you
are to evaluate
blood
cross-contamination
only, we can use
healthy
volunteers, the
number which has
to be determined
statistically.
Then we can use
the same
protocol, saline
injection and
then collect the
data after every
single use.
And I want to
make a note
here. If you are
using this
device between
users, we are to
sterilize the
device.
And then the
second thing is
if you want to
look at the
potential for
HBV
transmission, we
have to change
the population
now. We have to
think HBV
positive
volunteers, but
we can follow
the same
protocol.
So these
procedures,
actually, that I
haven't proposed
this but this
has been
discussed before
by, for example,
Dr. Bruce
Weniger at CDC.
He discussed
this at Global
Vaccine Research
Forum in 2004 in
Switzerland.
So this is all
of the aspects
that we have to
think about when
you develop a
strategy. So I
want to stress
the points
again. What are
the constraints
for developing a
safety
evaluation
strategy? There
are many
unknowns. Only
animal studies,
no clinical
studies other
epidemiology
studies. But if
you look at the
proposed number
of HBV copies
required for
transmission,
there's 10
picoliter maybe
inaccurate. So
we have to
realize what is
the lowest limit
of detection
that we want to
achieve.
Then other test
methods
available to
achieve the
required limit
of detection,
there are test
methods but what
is the limit?
That is the
thing that we
need to look at.
Then, again, one
other point I
want to stress
here is that
impact of
dilution factor
that has to be
accounted for.
If picoliter
blood is the one
that can
transmit the HBV,
can this be
measured
correctly when
diluted in the
ejected, that is
one that goes
into the device.
So these are all
the questions to
ask yet.
So coming to the
summary -- next
slide, please.
So this
presentation
summarizes the
current
available
methods that can
be used to
assist the
safety of MUNJI
devices. And we
have HBV model,
it's a good HBV
model for
evaluating the
contamination.
And there are
test methods
available, but
none of these
test methods are
validated. And
then we don't
know what the
transmission
limit we need to
look at.
So based on this
it is not clear
that these
methods can be
applied to the
investigation of
potential
cross-contamination
by MUNJI
devices.
Thank you.
CHAIRMAN
EDMISTON: Thank
you very much.
MR. LIPMAN: All
right. That
brings us to our
panel questions.
The first is
identify the
scientific
questions that
need to be
addressed to
demonstrate
whether MUNJI
devices are safe
for multiple
patient use in
the United
States.
Second, discuss
the adequacy and
feasibility of
the currently
available
methods to
assess the
potential for
cross-contamination
and the risk of
disease
transmission by
MUNJI devices.
The third,
Feinman, et.al.
in 1984
suggested that a
volume of blood
as small as 10
picoliters can
transmit
hepatitis B
virus in
chimpanzees.
However, this
finding is based
on a single
animal study.
Considering the
potential public
health benefit
of MUNJIs is
there a
threshold volume
of blood
contamination
that presents an
acceptable risk?
If so, what
threshold would
be considered
acceptable?
CHAIRMAN
EDMISTON: These
questions will
be part of the
Panel
deliberation
this afternoon,
and they will be
repeated again.
Before I go any
further, I'd
like to ask for
clarification
from Dr. Lin. In
our discussion
as we go through
this this
afternoon are we
addressing those
pre-amendment
devices that are
currently in
circulation or
are we
considering
answers to
questions that
will be
incorporated
into future
guidance
documentation?
DR. LIN: I think
that the answer
is both. As you
know, the
pre-amendment
device legally
it is still
considered
legally
marketable.
Every presenter
also mentioned
that we also has
clear some of
those MUNJI
device after
1972. So when in
your discussion
you have to
consider all
those potential
-- all those
legally mandated
device. So that
discussion will
be built into
our guidance
document in this
area.
CHAIRMAN
EDMISTON: At
this time I'd
like to invite
the members of
the Panel to
address any
questions that
they might have
to the
presenters from
the FDA. Yes,
Dr. Word?
DR. WORD: I have
a few questions.
One, if you're
looking for new
indications or
seeking new, I
guess, MUNJIs,
or that they're
all referred to
that; are you
looking to
utilize them in
one segment of
the population
or the entire
population?
I guess my
question because
I come from a
pediatric
background. Are
you saying do
you want it for
everyone or do
you want it just
for adults?
MR. WATSON:
Actually, I
think the answer
would be any
suggestions you
wold have in
that are would
be helpful.
Right now
they're
generally used.
There's no
restriction on
pediatric or
adult. The
assumption is
any appropriate
patient that can
be used for that
vaccine this
device can be
used on that
patient.
So if you have
suggestions
about that,
maybe you think
there's a
population that
is best suited
for this, we'd
be grateful if
you'd offer that
suggestion. But
to answer your
question, right
now they're
generally used.
There's no
restriction
whatsoever on
who these
devices can be
used for.
DR. WORD: I
guess the next
question I had
was when you
looked at safety
with your
chimpanzee data,
you talked about
I think it was
10 picoliters
were considered
acceptable?
Anything below
that would be
acceptable. But
yet you stated
also that it was
known to
transmit
hepatitis B even
if you went
below that. And
so I didn't
quite understand
how that number
10 came about.
And the reason I
say that because
if you're
looking at using
it in a
population,
we've had
universal
hepatitis B
immunizations
for the last 13
years. So we
have all
children up to
13 and we've had
catch-up, and we
don't have
others. And as
one of the
public speakers,
we don't have
hepatitis C
that's routinely
done.
And I guess my
question, and I
don't really
know what the
obstetricians
do, I don't know
if they
routinely screen
for hepatitis C.
I know they do
hepatitis B, and
they may not do
hepatitis C
routinely. I
don't think they
do. And if
that's the case,
then that may
not even answer
the question if
you're talking
about using
MUNJIs. You
might talk about
hepatitis B, but
still doesn't
address
hepatitis C.
MR. WATSON:
Right. I think I
might defer that
question to Dr.
Bezabeh.
My understanding
of it is
hepatitis B is
the most
virulent of the
strains and
that's why we
were looking at
hepatitis B. But
I'll leave that
up to Dr.
Bezabeh.
DR. BEZABEH:
Yes. What Tony
said was right,
you know. People
have looked at
hepatitis B
virus because it
was the most
high infectious
and it's easy to
measure.
The 10
picoliters was
from one study
in 1984 trying
to measure the
minimum amount
of blood that
can transmit
infectious
particles. And
serial
dilutions, they
have it right at
10 picoliters.
But to our
knowledge
there's no safe
limit, accepted
safe limit that
would be safely
transmit between
injection
devices. And
that's why one
of our questions
is because what
is an acceptable
safe limit of
blood?
CHAIRMAN
EDMISTON: Are
there any other
questions from
the Panel
members? Ms.
Petersen? Mr.
Layton?
DR. LAYTON: Yes,
I have a couple
of questions.
The first is
relative to the
intended use of
the device. Are
there separate
-- are any of
these devices,
or are there
separate
indications
depending on the
use with respect
to intradermal,
intramuscular or
subcutaneous or
can the same
device be used
for all
injections?
MR. LIPMAN: We
do usually have
different
testing for
those different
indications.
Basically, that
would be based
on the depth of
penetration and
the ability to
get to the
desired tissue
that the
injector is
indicated for.
DR. LAYTON: So
there are
different
standards from
that
perspective?
MR. LIPMAN:
Right.
DR. LAYTON:
Thank you.
The second
question goes
back to the 2004
WHO
International
Conference. Did
they recommend a
particular test
method? I missed
that if that was
-- they did not?
They recommended
studies, but not
a particular
test method.
Thank you.
CHAIRMAN
EDMISTON: Dr.
Arduino?
DR. ARDUINO:
Mine go along
with whether
it's intradermal
or subcutaneous,
whatever,
intramuscular.
For each jet
injector are
there different
settings that
you could set or
are they
separate
devices?
MR. LIPMAN:
We've actually
reviewed devices
that have -- I
mean, there are
a variety of
means to deliver
at different
depths. I'm
familiar with
different size
orifi, orifices,
whatever the
word is.
Different
injection
techniques
potentially -- I
don't
particularly
know how
accurate the
method is, but
pinching the
skin to actually
attempt to
create more
tissue to inject
into versus, you
know, letting
the injector
just inject
directly into
the skin to
reach, say, an
intramuscular
injection versus
a subcutaneous
injection.
Does that kind
of --
DR. ARDUINO:
Yes.
CHAIRMAN
EDMISTON: Dr.
Word?
DR. WORD: Just a
question related
to, say, let's
say if these
MUNJIs were
available, one
of the things
when you looked
at the adverse
effects, because
when you came up
with the swine
flu, one of the
things that
crossed my mind
immediately is
that I don't
know what my
mother received,
but I know she
told me she
thought her arm
fell off when
she got her flu
vaccine in '76.
And I don't know
if they used one
of those. But if
you're dealing
with adverse
effects and if
you're saying
you're looking
at who is
administering
them, because
you're going to
have some of
that
variability. So
I'm wondering I
don't know how
you control for
that.
I mean, I can
control for it,
but easier with
an injection.
And with the
other, I'm just
not sure how do
you control for
that or have you
thought about
how you control?
Or when you
talked about
contamination,
how often do you
check to see if
there's blood in
there?
MR. LIPMAN: The
users of these
devices would
definitely
ensure that at
least by visual
examination that
there is no
blood remaining
on the tip of
the injector.
But I mean
there's the
potential for it
to get back into
the fluid path.
You can't always
visually
identify that
there's any
presence of
contamination
present. And
that's kind of
the issue.
CHAIRMAN
EDMISTON: Dr.
David, do you
have any
questions?
MR. DAVID: Yes.
I have three
questions. One
relating to
previously asked
on the intended
use, and mostly
on the
definition that
you give the
MUNJIs. And my
question would
be why not look
at some of the
cross-contamination
principles and
look at the
device
definition by
the way of
possible contact
with the skins
exist. For
example, devices
that might have
continuous jet
flow, devices
that might have
various distance
gap producing
mechanism,
etcetera,
etcetera. And
that would
allow, perhaps,
some better
design of
devices and
validation of
their
performance
because you're
preventing
cross-contamination
to begin with.
So that's one
question.
MR. LIPMAN: We
actually have
representatives
here from a
company who is
having to design
based on those
ideas exactly.
Felton
International is
present here,
and they will
probably talk a
little more in
detail about the
testing they've
done on their
jet injector.
But, I mean,
they do actually
create a gap.
They have
disposal skin
contacting
device you have
to inject
through certain
layers to
actually get to
the body; the
idea being that
it would be much
more difficult
for any of the
stream or blood
to get back up
through that
small orifice
that's created
by the jet and
into the fluid
path.
So they have
attempted to
minimize it, but
the question
still remains
how can we
evaluate whether
or not they have
mitigated that
risk
sufficiently.
MR. DAVID: My
second question
relating to your
conclusion about
the single MDR
report that
cross-contamination
was result of
improper use.
And since we are
reviewing what
is considered
proper use, I
wonder had you
reached that
conclusion?
MR. LIPMAN: It
actually wasn't
even a MUNJI
device. It was a
device that had
-- actually it
may have been a
MUNJI device.
But either way,
it was supposed
to be used for
one person only
and then either
sterilized or
replace the
fluid contacting
components. But
instead, the
device was
actually used
for five patient
consecutively,
and that wasn't
the way it was
supposed to have
been done.
MR. DAVID: I
will go back to
my first
question that
I'm not sure
that the
definition of
low load and
high users is
appropriate.
MR. WATSON: I'm
sorry. We have
some more
information on
that last
comment.
DR. BEZABEH:
Just to clarify
the MDR report.
It did not
document any
closed
contamination.
There was just
misuse. So there
was no
documented
transmission or
infectional
cross-contamination.
MR. DAVID: My
third question
is about the
effort that FDA
put into looking
historically
since it was
noted here that
the DoD has
significant
amount of data
use of MUNJIs,
what are the
effort the FDA
puts to review
that source of
data use of
MUNJIs?
MR. WATSON: We
primarily looked
at what was out
there in the
literature that
the DoD had
published.
We haven't
actually
received
anything
directly from
DoD regarding
safety
information
about MUNJIs.
Most of -- well,
whatever the DoD
wants people to
know is out
there in the
published
literature.
Whatever other
information is
available, may
or may not be
available to FDA
directly. So
we've primarily
looked at what's
in the public
domain.
CHAIRMAN
EDMISTON: Any
other questions
from the Panel
members? Dr.
Lin?
DR. LIN: If I
may, just to add
to FDA's
comment. I think
probably for the
Panel members
probably need to
be recognized
that this is a
510(k) device.
It's close to
510(k) device
and I think that
the previous
presenter has
mentioned that
we are talking
about
substantial
equivalence;
that means that
you'll compare
the new device
with the current
market device.
You can even
compare with a
pre-amendment
that earlier,
like in 1950
something, those
device. That's,
as I mentioned
before, is still
considered
legally marketed
device.
So now when we
compare so
called
substantial
equivalence,
that means that
the manufacturer
would have to
establish that
they are as
safe, as
effective as
those legally
market device we
call predicate
device.
So that's the
concept how we
so call create
this device for
marketing. And
now that the
question is what
is considered
the criteria to
establish a safe
as effective,
that's the
issue. That's
what we try to
address. Because
the science
changed when we
review, like
early in the
'80s or '90s as
compared to now.
The emphasis is
quite different,
particularly for
in person
disease
prevention
control, quite
different. So
that when you
discuss the
FDA's question,
please keep that
in mind.
And then second
comment I wanted
to help with
that, I think
Dr. Word, you
mentioned about
the use and how
FDA treats the
users
participating.
That's most of
the time when we
do reviewing, we
will look at the
user's
instruction or
labeling. And
that is also the
area we would
like to hear
your input, too.
When we create a
guidance
document, what
kind of
information we
need to ask
manufacturers to
clearly indicate
in their
labeling that we
would appreciate
your input in
that regard.
Thank you.
CHAIRMAN
EDMISTON: Now
your statement
about
equivalence
really relates
to the delivery
of an effective
vaccine dose or
whatever you're
delivering. It's
not addressing
the concept of
infectivity or
safety from that
perspective,
correct? But as
the last
surviving member
of that 1999
Panel, it looks
like we have a
lot more data
available to us
for
consideration
than we had six
years ago. And
the question
that I rally
have is, you
know Mr. Hooks'
presentation was
compelling.
However, it was
anecdotal to the
point that we
don't have any
real evidence
relative to
risk.
And I suspect my
question is with
the devices that
are currently in
place, as any
assessment been
made in terms of
the relative
risk associated
with the use of
these devices in
acquiring an
infective dose
of whether it's
hepatitis C,
hepatitis B or
HIV?
MR. WATSON:
Shewit, what do
you think about
this question?
Is this a
question that
you might be
able to answer?
I just want to
make sure I
understand your
question. Are
you asking about
the
effectiveness of
actually
delivering --
CHAIRMAN
EDMISTON: No,
not at all.
MR. WATSON:
Okay.
CHAIRMAN
EDMISTON: What
we're talking
about now,
because I think
that's the issue
that we have to
separate here.
We're not really
concerned about
to a great
degree the
effectiveness of
delivering an
appropriate dose
of the vaccine.
What we're
concerned with
is how effective
is the device at
preventing the
transmission
cross-contamination
of an infectious
entity.
So my question
is relative to
'99 when the
committee
requested a some
postmarket
surveillance be
done, has any
consideration
been given with
the devices
currently in
place what is
the relative
risk of
acquiring an
infectious agent
with the current
device in place
without
realizing that
to a great
degree the risk
is associated
with the
compliance and
how the device
is being used?
So has any
consideration
been made of
what this
relative risk
might be?
MR. WATSON: I
think Dr.
Michaud might
have an answer
for us here on
that one?
DR. MICHAUD:
Ginette Michaud,
Deputy Division
Director of
DAGID.
I think it's
very hard to
answer your
question. The
reason we're
here today is to
get advice from
and
recommendations
from the
panelists as to
how we should
best assess the
risk of
cross-contamination
due to MUNJI
devices, or that
potential risk.
And so it's very
hard not knowing
the answer to
that how would
we determine the
relative risk as
compared to the
earlier designs
of these
devices.
CHAIRMAN
EDMISTON: I
appreciate that
comment. And
this goes back
to my first
question to Dr.
Lin. So
therefore our
deliberation
will have a
profound effect
on devices
currently in
place?
DR. LIN: Right.
CHAIRMAN
EDMISTON: All
right.
Yes, Dr. Word?
DR. WORD:
Perhaps you
stated this and
maybe I don't
recall. How many
devices are
actually being
utilized?
Because when I
looked at it,
you said that
there were a
number of -- you
know, CDC
recommended it
only for risk,
you weigh the
risk and
benefits. WHO
doesn't utilize
it. And I'm not
really concerned
about their use,
because it
doesn't effect
the United
States right
now. I know the
impact that we
have will
eventually have
a global effect,
whatever
recommendations
comes from here.
How many devices
are actually
being utilized?
MR. LIPMAN: I
can't speak
precisely to the
number of
devices that are
being marketed.
I can tell you
what I'm
familiar with.
We have cleared
two dental
devices for
delivering
anesthetic
during dental
procedures that
are MUNJIs. We
haver four
cleared, at
least four
cleared MUNJIs
for mass
immunization
intended use. Of
those four,
there are most
likely I'm
thinking two
that are
probably --
since there are
actually, you
know, these WHO
and CDC policies
against using
these devices,
the
manufacturers
have, obviously,
had a very
difficult time
marketing their
devices within
the United
States and the
world. So I
think Felton
actually may be
able to give you
a better idea
for how many
devices are
actually being
used and whether
they've been
able to market
their device.
MR. WATSON: One
thing to keep in
mind is that
even though
these products
may not be
widely used
anymore, we're
still getting
submissions for
them. And to the
extent that we
have to evaluate
them, we would
like some input
on what you
think we should
be doing here.
Because we're
still a
clearinghouse
for the world,
the FDA. So even
though they may
not be
necessarily
marketed here,
companies will
come to the FDA
to get clearance
because the idea
is get clearance
and FDA and a
lot of the rest
of the world
will accept
that. And we
would like to be
certain that
whatever we're
clearing is
something that
we consider
clinically
acceptable here,
not just based
on previous
standards for
clearing these
products.
So the actual
number, we don't
really know
that. We don't
really have
records for that
here. But we do
know we get
asked to clear
them. So that's
sort of one of
our concerns.
CHAIRMAN
EDMISTON: Dr.
Butcher, do you
have a question?
DR. BUTCHER:
It's been
answered.
CHAIRMAN
EDMISTON: Are
there any
further
questions by any
members of the
Committee?
I think we'll
move on to our
next
presentation
from Dr. Martin
Friede from the
World Health
Organization.
DR. FRIEDE:
Well, thank you
very much for
inviting me to
attend this. And
I would like to
reiterate
something that
was said a few
moments ago. The
recommendations
from this Panel
will have a
global effect.
So, first, I'd
like to
apologize. I
have modified my
slides slightly
compared to what
you received.
And this is
because I
learned last
night that Dr.
Weniger was not
able to attend.
So I have added
some more
background
slides. So I
hope this does
not effect what
you have too
much, but there
is some more
data.
So if we'd
please go into
the first slide.
So we've already
heard quite a
lot of
background about
the early
history of
safety concerns.
And,
unfortunately,
my eyes are
getting worse
and worse with
age. I can
hardly read that
myself. But
let's go through
this.
If we begin
around about
1959 there
was already an
evaluation done
using precipitin
test for human
serum. And this
really showed up
negative. And
this group in
1959 were also
unable to
transfer hog
cholera from one
viremic pig to
another. So this
was really the
beginning of the
evaluation of
safety.
In 1962, though,
Eli Lilly &
Company, I'll
show you this in
a moment, but on
their inference
of product
insert, that
bleeding could
occur and that
this would carry
a risk of
hepatitis, and
that it
recommended to
the doctor that
if blood was
observed, then
resterilization
should be done.
1970 bleeding
was noted on the
nozzles, on the
skin and blood
on the nozzles.
And it was
hypothesized
that disease
transmission
could occur.
And in another
1970 paper there
was an increased
detection of
albumin on
nozzles.
And 1981 there
was a study
done, this time
negative, no
hepatitis B
surface antigen
detected by
radioimmunoassay
after injection
of just two
volunteers, both
of whom were
hepatitis B
carrier
patients.
Next slide,
please.
So I certainly
can't read this,
but I know
what's written
there. This is
the product
insert from the
1962 package
from Eli Lilly
and it states
somewhere there
under red lined
that if bleeding
does occur, and
bleeding does
occur sometimes
with jet
injection, then
the nozzle
should be
resterilized. So
there was
recognition then
that hepatitis B
transmission
could take
place.
Next slide,
please.
Well, I think
the change to
the world jet
injection took
place in 1985.
And in 1985
suddenly we had
evidence of
risk. This was
the very well
known case of
the weight loss
clinic in
California where
a hepatitis B
outbreak took
place. But I
would like to
emphasize, this
is a fairly
unique
situation. These
were people
coming back time
after time. I
believe it was
15 to 30 times
over a two
months period.
Back to the same
clinic where
they were being
injected in the
same small
population where
there must have
been one high
titer carrier
that was there
who could
reinfect this
population.
Also, one this
one single
device and was
this device
being properly
used. So this
opens up a lot
of questions of
how to ensure
that devices
that may appear
to be safe, how
do we ensure
that they are
being properly
used and how do
we ensure that
they are
retaining their
safety over time
and in the hands
of everybody?
Next slide,
please.
This is some
data taken from
that California
study. Printed
in the Morbidity
Weekly Report.
And this shows
that when the
jet injector was
no longer being
used in that
clinic, we began
to see a decline
over the next
several weeks of
hepatitis B
onset. So this
was really the
proof. But we
must recognize
that this is not
quite the same
situation as
immunization
where you
typically go and
get one
injection, maybe
once per year.
Next slide,
please.
So after 1985
the world
changed
slightly, and
suddenly people
really began to
look at what
were the risks
of using these.
And, again, I'm
stretching my
eyes to see
this.
1985 there was a
demonstration
done that the
LDH virus, this
is a mouse
lactic
dehydrogenase
virus, could be
transmitted
experimentally
between mice
using a jet
injector. Again,
a comment here.
How does the
thickness of the
skin of a mouse
represent a
model for human
beings? And if
you were to give
a mouse a jet
injection with
an injected aim
to give
intramuscular
injections, this
would probably
cause a
tremendous
damage to the
mouse.
1980 hepatitis B
was found on the
skin on the site
of injection,
however it was
not found on the
nozzles of the
injection.
1994 this was a
study by Mr.
Brito. Blood
detection in the
ejectates. So
the volunteers
were injected
and then the
next shot was p
ut into a tube.
And they were
using the
forensic occult
blood detection
stripes which
measure about
2,000 picoliters
as limit of
detection. And
in roughly one
percent of the
ejectates, blood
was detected.
Now this
introduces the
concept of
picoliters.
We've already
heard brought up
this concept 10
picoliters is
the minimum
level of blood
that can
transmit
infection. I
hope that in my
presentation I
will show you
that this is not
a scientifically
sound
observation, but
we will see how
we can address
this.
So already at
2,000 picoliters,
one percent of
the ejectates
did have blood
in them.
1997 a VEE virus
was transmitted
between animals
using three
Russian jet
injectors, one
of which I
understand is
the originator
of the Felton
device which has
subsequently
been prior
approved.
1997 a very
interesting
paper published
from Bulgaria,
Dimache, et.al.,
this was in
Vaccine. Now
this is
interesting
because no
hepatitis B
transmission was
observed in
population. So
this is a field
study. 38,000
intradermal
injections were
given with a
disposable
spacer, which
they claim was
something like a
protection cap.
And this is very
interesting.
We'll discuss
this in a moment
as to what this
does not mean.
2001, this has
already been
mentioned, a
meta analysis of
hepatitis B in
Brazil showed
that people who
had received the
yellow fever
vaccine via a
jet injector
were much more
likely to have
also been
infected by
hepatitis B.
Then two studies
that I will
briefly discuss
have already
been discussed.
2001 the calf
model, serum
albumin was
detected in the
ejectates, and
that's an
unpublished data
already
discussed about
a clinical
model.
Next slide,
please.
So in the calf
model what was
done here is
that four
different
injectors were
used and saline
was injected
into the calves
and then
injected into a
tube. And using
a calf or a
bovine serum
albumin assay
looking to see
what was taking
place. What is
important out of
this is that you
see that there
are a lot of
samples that
have between 10
and 50
picoliters and
quite a lot that
have between 50
and 1,000
picoliters of
serum albumin.
So this shows
that all four of
the old model
jet injectors
were
transmitting
quite often
quite a
significant
amount of blood.
Certainly what
we would
consider to be
an infectious
level of blood.
Next slide,
please.
Now this is
unpublished
data. Again,
coming from
Brazil using,
again, old model
injectors. And
when I refer to
"old model
injectors," I am
comparing this
against improved
injectors that
may be available
soon.
What was done
here saline was
injected into
the volunteers
and then three
injections were
made
sequentially
into a tube. And
using a human
serum albumin
study looked at
how much blood
was there. Now
this study had,
apparently, a
limit of
detection of 10
picoliters. So
wherever you see
something
positive, it
simply means
greater than 10
picoliters.
And what we're
seeing here is
whether it was
wiping with the
nozzle or wiping
without the
nozzle, we had
between 7 and 11
percent of the
ejectates were
contaminated
with blood.
However, what
was also done in
this study was
injecting saline
into the tubes
before injecting
people. And you
see there are
positives there.
So this really
begins to
question this
assay. We were
getting false
positives here.
And I will
discuss this
later. But the
reliability of
this assay is
doubtful.
Next slide,
please.
So what has been
the reaction of
the public
health
organizations?
First of all,
we've had over 2
billion
immunizations
given worldwide
from 1952 to
1990. We've had
warnings on the
risk of blood
transmission.
We've had the
hepatitis B
outbreak. So in
1987 WHO
recommended
restricted use
of these
devices. And
finally in 1996
we actually
recommended
against the use
of these
devices.
And from 2000 to
now there has
been the
development of
new generation
devices aimed at
overcoming these
safety concerns.
Next slide,
please.
So I'd like to
summarize and
the rest of the
meeting
summarizing a
meeting that we
had in March
2004 which was
aimed
specifically at
determining the
safety of these
new generation
devices. And by
"new generation
devices" I mean
devices that are
aimed at
overcoming these
safety concerns;
that have a
built in safety
device.
The questions
are how
infectious is
blood? How do we
measure it? How
do you model the
risk? What level
of risk is
acceptable? And
our conclusions.
Next slide,
please.
So this is, I
think, possibly
my most
important slide,
is how
infectious can
blood be? We've
already heard
that hepatitis B
is far more
infectious than
hepatitis C,
which is more
infectious than
HIV. And there
is a CDC
reference for
this. Now we've
heard the
statement that
10 picoliters is
able to transmit
infection to a
chimp. This
comes from the
Bond, et.al.
paper 1984. Ten
picoliters could
infect one
picoliter could
not. However,
this was one
study on one
sample. So it
means for that
sample of serum
had that type of
viremia, 10
picoliters was
able to, one
picoliter was
not. And that's
all that means.
So at the
meeting last
year we tried to
answer the
question of how
infected is
hepatitis B. And
you see over on
the right hand
side a graph
which is taken
from the Lindh
paper. And this
shows two lines.
The upper line
are people who
are HBE positive
with the HBE
antigen. And it
shows their
viremia in terms
of genome
equivalence per
milliliter. The
average is
around about 10
to the 9. It
goes up to 10 to
the 11. However,
we also heard at
the meeting last
year that in
rare cases when
people have both
HIV and
hepatitis B,
viremia can go
up far, far
higher; 10 to
the 12, 10 to
the 15 even.
So for the rest
of this
discussion I
have just
assumed that 10
to the 9 is an
average amongst
these HBE
positive
carriers. And we
have done a bit
of modeling and
assumed that
hepatitis B
carriers, of
these 20 percent
have high
viremia, and
this 10 to the
9.
And the
conclusion of
this is that a
fraction of a
picoliter can
transmit
infection. So if
you haver a
viremia of 10 to
the 9, this
means you have
one genome
equivalent per
picoliter. But
there's a
probability, of
course, that you
may have more
than one genome
equivalent per
picoliter
because you
never know how
these things are
being
distributed. And
also you may run
into somebody
who has a
viremia of 10 to
the 15, in which
case one
picoliter may
have a very high
number of genome
equivalence.
So the next
slide, please.
Because of the
recognition that
we have to go
below 10
picoliters, we
were looking at
assays to
measure blood
contamination.
Now the human
serum albumin
assay had been
developed as a
surrogate
marker. Since
human serum
albumin is the
main protein
component within
blood, it was
felt that this
was a good
target to be
going for to
measure how much
blood could be
on the nozzle.
This was
developed by
Kings College in
London.
And an improved
assay was
developed by
them where they
claimed they
could develop,
approximately
they could
detect
approximately
three picoliters.
That was limited
quantification,
limited
detection, about
one picoliter.
However, as has
been already
mentioned, serum
albumin is
everywhere. It's
in our spittle,
it's on our
skin, it's in
our hair. And
for example,
dead skin may
not have any
probability of
transmitting
infection, but
it will give you
a positive
single. So this
presents a lot
of problem using
the human serum
albumin assay as
a surrogate
marker for
blood.
Also, when WHO
sent this assay
out to two
independent
laboratories, we
discovered that
you could not
validate this
assay and it was
the independent
laboratories
gave limited
protection or
limited
quantification
between 15 and
30 picoliters.
So it was
therefore a
requirement for
a more reliable
and more
sensitive assay.
Ideally, we need
to be able to
really measure
infectivity.
Measuring blood
volume, per se,
doesn't tell you
much. So we felt
that a PCR
analysis of, for
example, the
hepatitis B
virus from
highly viremic
carriers, this
gives you an
idea of really
how much, what's
the probability
of getting
infected.
Next slide,
please.
Here this shows
the comparisons.
When the Eli
Lilly product
insert said if
you see visible
blood,
resterilize it,
that's about 0.1
microliters.
There's a limit
of what you can
see. Chemical
blood tests is
about .01
microliters.
Measuring
surface antigen
with an analyte
is about .001
microliters. The
albumin assay,
15 to 30
picoliters. And
we believe that
using modern
techniques you
can detect
hepatitis B
virus at about 3
genome
equivalents. So
this would be
about 3
picoliters of
that high titer
serum.
So we then tried
to -- since we
accept that you
can get disease,
you can get
disease
transmission
with less than
10 picoliters,
the question is
how do you model
this risk? We
have to have an
idea of what
risk is there
with one
picoliter. What
risk is there
with .1
picoliters? And
this begins off
with the
assumption that
risk of getting
hepatitis B
virus is
proportionate to
the endemicity.
It's logical. If
you are in a
room where 50
percent of the
people in the
room are
hepatitis B
carriers and you
will be
receiving a jet
injection
subsequent to
one of them, you
have a higher
probability than
if you're in a
room where there
is only 1 per
1,000 with this.
So that's
logical.
In the USA you
have less than 2
percent carriers
This is WHO
figures. In
Africa,
Sub-Sierra in
Africa, there
are between 8 to
20 percent of
the population
that are
hepatitis B
carriers.
So let's go into
some very rough
modeling. This
was presented at
the meeting last
year. And I've
just tried to
summarize this
taking one or
two examples.
If, this is a
very big if, if
each injection
transmitted .5
picoliters, now
this would be
safe by our PCR
assay that I
just discussed
which is
measuring about
3 genome
equivalents. So
.5 picoliters
would pick up
nothing. We
would say safe.
If 2 percent of
the population
were carriers
and if 20
percent of these
were HBE antigen
positive, in
other words high
titer carriers,
then also if one
ID50 was 10
genome
equivalence -- I
should mentioned
that WHO tried
to find out from
hepatitis B
experts what is
the ID50. How
many genome
equivalents does
it take to
transmit
infection?
We heard from
Bob Purcell, not
at the meeting.
This was by oral
communication he
gave us. That 10
genome
equivalence may
do it. We heard
other experts
said maybe a 100
genome
equivalence do
it. So we don't
really know. But
we're taking a
worse case
scenario and say
10 genome
equivalence
could transmit
infection.
Now, we can do
some mathematics
on this. And
this would say
that on a
population with
10 to the 9
genome
equivalence per
millimeter,
which is your
high titer
carrier, one ID5
would be 10
picoliters. But
what happens if
you are giving
less than 10
picoliters? So
we worked out a
mathematical
formula which
tries to express
the fact that
this is not a
linear decrease,
but we expressed
the probability
that N ID50s
give you an
infection as
being one minus,
not 25 to the N.
You could also
get roughly the
same number by
just dividing
the number of
microliters or
picoliters that
are being given
by the 10
picoliter
sample, which
contains your
ID50.
In this case the
probability of
infection on
receiving .5
picoliters from
a higher viremic
carrier is .034.
And then to
calculate the
probability of
infection, you
have to work out
what is your
probability of
this person
being in line in
front of you,
which is the
probability of
having your
hepatitis B
carrier there
and the
probability that
that hepatitis B
carrier is a
highly viremic
carrier times by
the probability
of the .5
picoliters
carrying an
infectious dose.
And this comes
to -- you've got
the numbers
written there.
.000132, which
means that they
could be up to
-- and I
emphasize up to
132 infections
taking place per
million
injections.
However -- next
slide, please --
I'd like to
really show the
caveats of this.
First of all,
the ID50 that I
took there was
10 genome
equivalents.
This is the most
infectious that
we've heard
from. Other
scientists have
said it's more
like a 100. So
this would bring
us down to 12
infections per
million.
Now, first of
all, the studies
that have been
done including
the Bond study
in 1984, the
serum was
injected
intravenously.
Now when we give
jet injectors,
this is not
intravenously.
So it could be
that by giving
nonintravenous
delivery, we are
also going to
decrease the
infectivity by
not getting to
the blood, not
getting to the
liver. So this
could drop this
down even
further.
Also, there may
be other factors
such as drying.
The numbers that
we get there,
this is the
worst, worst,
worst situation,
the worst case
scenario
possible. It
assumes a linear
risk. It could
very well be
that below a
certain viral
load the risk
may be
infinitesimally
small. And it
also assumes
that every
ejectate is
contaminated. So
that is the
caveat for this
and it gives us
a number.
Now let's look
at risk
assessment in
field trials. I
already
discussed
briefly this
Dimache study
1997. This was a
slightly new
generation
injector. It had
a disposable
spacer. It was
not really a
protector cap.
38,000
injections were
given in adults.
This was in
Bulgaria where
the hepatitis B
endemicity is 5
percent. And
these volunteers
were followed up
for six months
to determine how
many cases of
hepatitis B
virus
infectivity took
place subsequent
to the
immunization,
which could be
ascribed to
cross
contamination.
And absolutely
none took place.
No observed
hepatitis B
infection in
vaccinees.
However, this
was a low volume
injector. It was
delivering .1 to
.2 mls
intradermally.
This is not the
same as the
studies we
talked about
previously which
were typically
intramuscular or
subcutaneous
with 0.5 ml.
Intradermally
one could
imagine a lower
splash back.
Zero out of
38,000 observed
infections. The
upper 95 percent
interval of this
is 4 per 38,000.
So we could be
having a risk of
really 1 per
10,000, risk of
infection and
still observe
zero to 38,000
in a field
trial.
So the field
trials to prove
safety would
require very
careful design
to give power.
And I think this
is one of the
real difficult
questions here.
When we're
dealing with
such low figures
of shall we say
10 per million
or a 100 per
million
infectivity as
being the
possible risk of
a device, how do
you see this
signal above the
background
noise?
If you go to
Sub-Sierra in
Africa where you
have a
background and a
high rate of
infectivity
taking place,
you have a high
noise. So how
would you see
your relatively
big signal. If
you do this in
the USA where
you have a
relatively low
background, you
will also have a
very relatively
low signal.
Determining your
signal to noise
ration in a
field evaluation
is going to be
exceptionally
difficult.
Next slide,
please.
So the
conclusions of
the WHO meeting
were that sub
picoliter levels
of blood can
transmit
disease. Ten
picoliters is
not a
scientifically
valid number.
Available blood
markers, which
were the serum
albumin, are
inadequate as
surrogate
markers of
safety. However,
PCR detection of
hepatitis B from
highly viremic
carriers is much
better.
It is feasible
to evaluate
safety for a
small sample
size by PCR.
However, how
does one take
into account
device aging and
device
misfunction? And
I'd like to
bring up a
question here
for your
consideration,
which is how do
you determine
the reliability
of the safety
mechanism? You
may prove that
your device is
safe in a small
trial of a 100
people, but how
do you determine
that the device
is reliable over
a long term?
This would
probably require
ex vivo and in
vitro studies,
but this will
have to be
considered.
We also
concluded that
it would be very
complex to
evaluate safety
for a large
sample size. So,
first of all,
going from this
small field
study using
highly viremic
carriers to the
population which
you're actually
using the device
in the
population, we
could not
determine the
ethical pathway
to get there.
Next slide,
please.
So, WHO
position. The
determination of
the safety of
MUNJIs is the
responsibility
of national
regulatory
agencies. WHO
will not
determine the
safety. This is
the
responsibility
of the national
regulatory
agencies.
Secondly, if
used property
needle injection
is safe. Now
this is a big
if. We know that
the injections
are not always
done properly
and we know that
disposal is not
always done
properly.
However, if done
properly it is
safe.
To the WHO is
not acceptable
to replace
injection by a
technology for
which the safety
is questionable.
So while we have
questions on the
safety, it is
not acceptable
to replace
needle and
syringe.
Needle-free
vaccine delivery
is desirable. We
recognize this.
If we can get
rid of needles
from the
immunization
program, this
would be
fantastic. Given
for the moment
the questions on
the safety of
MUNJIs, we
believe that
disposable
cartridge
injectors where
there is no
reusable path or
appropriately
safe
alternatives,
whether or not
they're cost
effective is
another issue.
We are
evaluating the
use of these for
vaccine
delivery.
Now, I'd like to
finish. Next
slide, please.
With two slides.
First of all,
these are the
points for
consideration.
We've already
heard about the
advantages:
There's no
sharps, there's
no waste, it's
fast and it is
low cost, very
low cost per
injection. The
comment to this
is that there
may be a risk.
Whether the risk
is a real risk,
whether it is a
risk that is
perceived by the
population, this
could really be
inhibitory of
these devices.
Daily cleaning
and
sterilization of
the fluid part
is required and
there may be a
risk if this is
not properly
performed. We
know that
ensuring the use
of syringes
properly is
difficult
ensuring safe
cleaning of
these multiuse
devices may be
complex. And we
also face the
problem of the
cost per device.
Under what
circumstances is
high speed
injection
required? It's
required really
where you have a
low ratio of
health care
worker to
population or
where you have
centralized, not
dispersed health
care.
And finally,
what level of
risk is
acceptable? So I
think we really
have to balance
the risk benefit
here. Needles
injection is not
always performed
safely. Needle
stick injuries
do occur. Needle
disposal is not
always performed
safely. However,
for the
individual, an
individual
receiving an
injection from a
sterile needle
and syringe runs
no risk. So we
have to look a
the risk to the
individual
compared to the
risk to the
population, and
I think that is
a question for
the Panel.
Thank you very
much.
CHAIRMAN
EDMISTON: Thank
you very much
for traveling to
Washington and
making this
presentation.
At this time
this
presentation is
open for any
consideration.
Do any members
of the Panel
have any
questions for
Dr. Friede? Yes,
Dr. David?
MR. DAVID: I
have two
questions. One
is relating to
the comment you
made about the
disposal
cartridge. What
do the study
looked at when
they looked at
this puzzle card
as far as
volumes and so
on?
DR. FRIEDE:
There has not
yet been a
study. We are
beginning to
evaluate these.
MR. DAVID: So
your statement
about it is an
alternative safe
is based on?
DR. FRIEDE: It's
simply because
there is no
reuse of the
fluid part,
there is no
reuse of the
nozzle. There
cannot be
transmission of
blood from a
nozzle because
the nozzle is
not reused.
The definition
of a MUNJI was
given
previously,
earlier on this
morning, as
being one where
the fluid part
is reused. In
the disposal
cartridge the
entire fluid
path, the entire
-- the whole
fluid path, the
whole nozzle is
used once and
cannot be
reused.
MR. DAVID: I
see. So the
whole fluid path
is replaceable
then, that's the
point?
DR. FRIEDE:
Completely.
MR. DAVID: Okay.
And if we can go
back to your
risk model
slide. Where was
it.
DR. FRIEDE: Next
one. That's
right.
MR. DAVID: Can
you just take me
again through
the ID50
argument.
DR. FRIEDE:
Okay. The one
figure that we
received from
Bob Purcell
suggested 10
genome
equivalents is
an ID50. So
let's just take
that as a
starting point.
Other people
have said 100.
Now, if you have
10 to the 9
genome
equivalence per
milliliter, this
means you have
one ID50 in 10
picoliters. In
other words, if
you receive 10
picoliters, you
have a 50
percent
probability of
becoming
infected by
definition of
the ID50.
So the question
is if you
receive less
than ten
picoliters, if
you receive one
picoliter, what
is the
probability in
one picoliter
that you are
going to have
ten genome
equivalents? So
this is our
applying the
statistical
laws.
You have a
random
distribution of
your ten genome
equivalents per
-- I'm sorry,
your 10 to 9 per
milliliter. What
is the
probability that
10 genome
equivalents are
going to be
found in one
picoliter?
MR. DAVID: Okay.
DR. FRIEDE: The
way to do this
is to use that
formula. Okay.
This is an
expediential
formula. So the
probability that
you will find an
ID50 in one
picoliter is
going to be 1
minus now .5 to
-- it's going to
be one divided
by 10.
MR. DAVID: So
you're making
actually two
arguments. One
is the volume of
the injected and
the other one is
the site
intramuscular or
intravascular as
two mechanisms?
DR. FRIEDE: The
caveat is this
concept of 10
genome
equivalents,
this comes
really from
intravenous
studies. And it
could very well
be. I'm putting
this as a
caveat, as a
scientist, that
when we deliver
this
intramuscular
and it doesn't
get straight
into the
capillaries or
into intravenous
system and go to
the liver, it
might take a far
number of genome
equivalents.
This is a worst
case scenario if
you take all
available data
that we have. So
we're really
looking at what
the worst number
could be.
MR. DAVID: Okay.
DR. FRIEDE: And
with that
number, you see
that your signal
is quite small
and it really
opens up the
question of how
would you see
this in a
population.
DR. ARDUINO: But
when we get to
risk, because
I'm doing some
stuff with
biodefense
stuff, an ID50
may not be
acceptable. What
happens when if
you shift the
curve and want
to look at an
ID10 or an ID1?
Well, your
number gets how
many -- you
know, it gets a
lot smaller,
doesn't it?
DR. FRIEDE: It
does. I put this
really as a
method of
looking at it.
Now those
numbers there
are not
validated
numbers. These
were numbers
that we put up
as a method of
approaching this
to enable you to
accept the fact
that 10
picoliters is
not a number, is
not suddenly
that below 10
picoliters
nothing happens.
Things can
happen below 10
picoliters. We
need to
determine what
is the worst
probability that
something will
happen?
CHAIRMAN
EDMISTON: Any
other questions
from the Panel
members? Dr.
Layton?
DR. LAYTON: Yes.
I have a
question on the
risk assessment,
the Rumanian
study where you
talked about the
lower splash
back risk than
0.5 ml
intramuscular
and you had a
question mark.
Would you care
to elaborate on
that relative to
this intradermal
versus
intramuscular
and any of your
observations or
knowledge
relative to the
knowledge
relative to the
level or degree
of splash back?
DR. FRIEDE: That
I put up -- we
have it on the
slide.
This is, as a
scientists, I
just imagined
that if you
inject .1 ml
intradermally,
you're going to
have a much
lower risk of
forcing body
fluids back up
onto the nozzle
than if you
inject a larger
volume deeper.
Intradermal
probably
shouldn't really
be giving you
any blood, and
there's not much
going in. The
volume coming
back is probably
going to be a
function of the
volume going in,
and also the
elasticity of
the tissue that
it's going into.
So I think
looking at this
study it's an
interesting
study, but as I
said there are
two caveats
here. One is
it's intradermal
and low volume.
And what I
really wanted to
bring this up
for is that
seeing zero in
this population
doesn't tell you
a lot.
DR. LAYTON:
Thank you.
CHAIRMAN
EDMISTON: Any
other questions?
I have a
question. From
your perspective
what troubles
you about these
devices? Is it
their design or
the hydraulics?
Because
obviously these
devices are not
going to go
away, especially
in a
circumstance
where we need
mass
immunizations.
DR. FRIEDE:
Okay. There's
two things that
worry us. And I
give you the
official point
of view here.
The first one
actually is to
do with the
maintenance of
these. That in
the populations
which are our
responsibility
to reintroduce a
cleaning
procedure which
has to be done,
and the
maintenance,
this is a very
big problem for
us.
The second
problem is that
while there is
concern of
safety, any
concern, for us
to impose on
countries to use
this device just
carries an
enormous risk
that until we
get really clear
evidence or a
clear consensus
that this is
safe, it is
going to be
difficult for us
to recommend to
countries to use
this. Because
any incident
that took place
would come back
and we would
struggle to say
we confident
that that
incident, your
infection, did
not occur
because of the
device. So until
then we are
standing by our
policy, which is
that
immunizations
will be given
with
auto-disabled
syringes. And
that the
auto-disabled
syringes will be
provided with
sharps disposal
boxes to try to
ensure that
sharps disposal
is done
correctly.
CHAIRMAN
EDMISTON: If
the devices are
used in a
compliant manner
the way they're
meant to be
used, do you
think the
devices are
safe?
DR. FRIEDE: The
devices that we
have seen
without a
protection cap,
we have data
from the calves
and the data
from the Hoffman
study in Brazil
to show that
frequent
contamination of
the ejected did
take place. And
that
contamination
was clearly of a
level of blood
that we are
convinced can
carry disease.
So the devices
which do not
have a
protection cap
which are to be
used for
giving
intramuscular
injection we are
convinced that
these carry a
significant
risk.
CHAIRMAN
EDMISTON: Okay.
Any other
questions by
members of the
Panel?
Well, thank you
very much for
your time.
I've been
informed that we
can do lunch.
Actually, we're
about half an
hour ahead,
which is
terrific.
I'd like to
invite you all
to lunch, and
we'll meet back
in one hour.
Is industry
going to be
making their
presentation? Is
12:00 fine for
industry
presentation? Is
everybody here.
Okay. Well the
plan at this
time is to
reconvene at
12:00 and begin
our industry
presentations.
Thank you.
(Whereupon, at
10:58 a.m. the
meeting was
adjourned, to
reconvene this
same day at
12:00 p.m.)
A-F-T-E-R-N-O-O-N
S-E-S-S-I-O-N
12:05 p.m.
CHAIRMAN
EDMISTON: I
would like to
now call the
meeting back to
order.
I'd like to
remind the
public observers
in the audience
that while this
portion of the
meeting is open
to observation,
public attendees
may not
participate
unless
specifically
requested to do
so by the Chair.
We will now
continue with
industry's
presentation
related to
today's topic.
And we have Mr.
Darin Lee
Zehrung, did I
pronounce your
name correctly?
DR. ZEHRUNG:
That's correct.
CHAIRMAN
EDMISTON: He
will be
addressing
Program for
Appropriate
Technology and
Health.
DR. ZEHRUNG:
Thank you.
Do I have to
make a conflict
of interest
statement at
this time?
CHAIRMAN
EDMISTON: Yes,
we would
appreciate that.
DR. ZEHRUNG:
Well, PATH is a
nonprofit
organization,
nongovernmental.
It is focused on
improving health
in the
developing
world. And we're
actually working
with a couple of
different
needle-free
injector
developers, one
of which is
Felton
International,
and that's the
technology that
I'll talk about
today. It's a
collaboration
with different
developers that
includes a
development
portion as well
as clinical
testing. But we
do not receive
any funds from
these
manufacturers,
and actually
we're
self-funded by
different
donors.
Next slide,
please.
So, as I said,
PATH is a
nonprofit
organization.
And this is our
mission: To
improve the
health of people
around the world
by advancing
technologies,
strengthening
systems and
encouraging
healthy
behaviors.
Actually, I'll
hold there.
We've actually
been involved in
the development
of safe
injection
technologies for
the past 20
years, either
disabled
syringes,
Uniject which is
a prefilled
injection
device, sharps
disposal
technologies all
focused on
improving
immunization
safety in the
developing
world. And I
work within a
program called
Technology
Solution within
PATH, which is
that is our
prime mission.
Next slide,
please.
So we talked
about this
earlier today.
What's the
technology need
for a high speed
needle-free
injector?
There's the
application for
mass
immunization
campaigns. In
the developing
world examples
are measles,
yellow fever,
meningitis and
there are other
examples. There
are also
emerging
vaccines that in
the development
pipeline that
could also be a
good application
for high speed,
high throughput,
high numbers of
injections for
those in the
developing world
such as meninge
which is focused
on West Africa,
malaria vaccines
and also human
papilloma virus
vaccines.
Pandemic
outbreak is also
another key
application for
this technology.
Influenza, you
know we've read
these recent
articles about
avian flu and
the potential
for outbreak.
There's really
not a technology
that exists that
could provide
high throughput,
mass
immunization to
those vulnerable
populations,
either in the
developing world
or in the United
States or
Europe, for that
matter.
Bioterrorism
response is also
another
important
application. And
I think that I'd
actually like to
hear from others
that represent
perhaps that
perspective to
see if this
technology or
what their plans
would be to
respond to an
outbreak or even
a bioterrorism
attack.
And then there's
the military
application.
Although we
heard about
earlier issues
with devices,
the first
generation MUNJI
devices, so to
speak, there
could perhaps
still be a need
for a high speed
injector in the
military.
Next slide.
So this is
actually a slide
that I received
from Dr. Bruce
Weniger, and
unfortunately he
could not be
here today. He's
actually Mr.
Needle-Free
Injector at CDC.
And I think he
has a very
prominent
position in the
needle-free
injector
community. And
he's done a lot
of work on
looking at the
efficacy of
needle-free
injectors in
delivering
multiple
antigens. So
this is a list.
And I think we
saw an earlier
version of this
in a
presentation
this morning
where there is
great historical
evidence, over
decades of use,
needle-free
injectors
delivering
different
vaccines.
Perhaps with the
new combination
vaccines and
newer vaccines
in development
there is not
this clinical
history, but
it's clear that
needle-free
injectors are
effective in
delivering
vaccines.
Next slide.
So this is a
technology that
we are
collaborating
with in terms of
Felton
International.
They're the
manufacturer.
And, actually,
if there are
more specific
questions about
the technology,
I would defer to
my colleague Dr.
Anatoly Loskutov
from Felton
International
who could
perhaps provide
more in depth
answers.
I'd like to
point out that
we see this
technology as a
design hybrid.
It's really not
a MUNJI. There
is a reusable
fluid path, yes,
but there's not
direct nozzle to
skin contact.
The key feature
of this
technology is
that it utilizes
a protector cap
as a disposable
shield. And
actually I've
passed around
samples of this
protector cap to
the Committee
members. This
shield is
intended to
prevent
cross-contamination.
And we've been
involved in
collaborating
with Felton over
the last several
years, a
combination of
in vitro and in
vivo testing to
build the safety
profile for this
technology to
demonstrate that
it does, indeed,
prevent
cross-contamination.
The current spec
for the device
is that it has a
fixed half cc
dose. It's
intended for
subcu delivery,
which most of
the developing
country mass
immunization
campaigns
deliver a half
cc subcu dose.
But with
different
orifice sizes
you could either
achieve an
intradermal dose
or
intramuscular.
We focused on
subcu for the
current specs
for the
technology.
And it's
hydraulically
powered. It does
not require
electrical
power. It
utilizes a foot
pedal and
hydraulics which
compressed a
spring within
the hand piece.
That provides
the energy then
to provide the
injection.
And we've
targeted in
terms of the
spec six
injections a
minute. Now,
it's not as
quick as the
earlier first
generation MUNJI
devices, but
it's more rapid
than needle and
syringe
delivery. So,
therefore, still
we would
consider it a
high workload
device.
It also requires
steam
sterilization of
a reusable path.
Let me point
this out here.
So this is the
hand piece here.
This is the
fluid path
portion. So
that's detached
from the hand
piece, cleaned
and then steam
sterilized.
And actually, I
think Jason
Lipman mentioned
this, there are
few
technologies,
MUNJI devices
that have
received 510(k)
clearance
post-amendment
era. And this
technology is
one of them.
Well, actually
last year in
2004 this
particular
design received
a special 510(k)
clearance based
upon an earlier
510(k) clearance
for a device
called the
BI-3M, which was
originally a
Russian design.
Dr. Loskutov
comes from the
original design
group, and
perhaps he could
talk about that
for those that
are interested.
Next slide.
So
unfortunately, I
had a video
demonstrating
the technology.
It doesn't work.
So what I'd like
to offer is for
the Committee
members that are
interested --
okay. Well, for
the Committee
members, I'd
like to offer I
could bring my
laptop to show
you the
operation of the
technology. And
then, again,
anyone from the
public
observing, if
you have
questions please
feel to contact
me or Dr.
Loskutov and
then we'll
demonstrate the
technology.
But basically
the protector
cap is placed on
the nozzle face.
Let me go to the
next slide. It
incorporates a
space between
the nozzle and
the injection
site. The
injection stream
passes through a
thin
polyethylene
film. And once
the injection
stream
penetrates that
film, that
enters into the
tissue. And any
splash back, any
contamination is
then contained
within the
protective cap.
And for the next
injection, the
protector cap is
discarded and a
new sterile
protector cap is
placed on the
nozzle face.
Another key
feature about
this protector
cap is that it's
auto-disabled.
Once you eject
it from the
nozzle face,
it's disabled so
that if you were
to put it back
on the injector,
you could not
provide an
injection
through that
spent protector
cap.
One key features
and perhaps
Anatoly could
speak to this
that's
development now
and it will be
available for
the next design
iteration, is an
interlock which
would require
placement of a
protector cap on
the nozzle face
for the device
to operate. So
perhaps, Jason,
you'll see that
in a subsequent
submission.
So in terms of
the benefits of
the technology,
a key feature:
Prevents
cross-contamination.
It uses a
protector cap.
And I think that
the PATH
position is that
we believe that
this technology
can be
demonstrated to
be safe. We
could talk about
the safety
design, we can
talk about
sample size, but
we have the
confidence that
this technology
could have great
application and
would be a safe
technology
eliminating
needles from use
in mass campaign
scenarios.
It's also high
speed, as we
talked about. It
allows for rapid
response.
One key feature
and one feature
benefit that we
see is that it
protects health
care workers.
There's no risk
to needle stick
injuries. And in
a mass campaign
when you're
dealing with
large numbers of
individuals, at
least in the
developing
world, we have
mountains of
sharps waste
that you need to
discard it. And
Dr. Friede had
mentioned that
the current
policy is to
bundle safe
injection boxes,
sharps waste
boxes, with
those
auto-disabled
syringes. But
there's still
the potential
for health care
worker needle
stick or for
community needle
stick injury in
terms of the
general public.
Many times these
syringes are
buried in a pit
behind a health
care center or
there's an
attempt to
incinerate them
or burn them.
Many times
unsuccessful. So
that there is a
general need, an
acute need, for
a needle-free
technology.
Next slide.
So, are main
focus in this
project has been
to conduct
safety testing
of the protector
cap injector,
the Felton
device. This
project has been
funded by the
Bill and Melinda
Gates
Foundation. He's
very interested
in this
technology for
mass
immunization.
And there have
been a number of
in vitro and in
vivo studies
that we've
conducted. What
I'm going to
present are our
recent studies.
There are
studies that we
have conducted
over the last
several years
that I won't
discuss today,
but if you're
interested I
could provide
that information
after the
meeting.
So fluorescein
testing as a
simple model. I
think earlier we
talked about the
challenges of
identifying an
appropriate
animal model.
Our focus has
been to focus on
a bench test
model using a
very sensitive
assay and marker
to demonstrate
that there's
cross-contamination
that does not
exist.
And then the
focus our human
safety testing
has been
hepatitis B
virus detection.
You know, from
the WHO meeting
that was held
last year, we
took that input
and we focused
on a method,
identifying a
method that
could be used to
detect hepatitis
B virus in
subsequent
injections.
Next slide.
So for the
fluorescein
safety testing
we use a very
highly
concentrated
fluorescein dye,
and that's a
surrogate for
high titer HBV
infection. And
the detection
limit of this
current approach
is .04 picoliter.
I think that
we've talked
about picoliters
and volumes of
infectivity
throughout the
meeting. And I
think that for
some it might be
a little
unclear, but
really what it
means is that
it's about 100
fold more
sensitive than
available PCR
methods.
The original
design of this
fluorescein test
focused on the
10 picoliter
threshold. But
given the input
last year at the
WHO meeting, we
put that aside
and just focused
on if anything
could be
detected with
the method, then
that would be
the definition
of
contamination.
So I think that
the current
results that I
can show you
demonstrate that
with the
protector cap
injector there's
no
cross-contamination
in comparison to
predicate
devices such as
earlier MUNJI
devices there is
demonstrated
cross-contamination.
The samples that
are generated in
the PATH
laboratory are
sent to a third
party
laboratory, MDS
Pharma in the
base outside of
Seattle,
Washington. And
they use their
equipment to
analyze samples.
Next slide.
So thanks Dr.
Friede, he gave
me this slide
earlier today.
So I would like
to stress my
appreciation for
this.
What I want to
point out is
that in
comparison to
the other
contamination
assays that Dr.
Friede had
presented, the
fluorescein
assay really
exceeds the PCR
methods in terms
of a detection
limit. So it's
very sensitive,
it's very
specific in
terms of an
assay. And we
believe a good
surrogate aside
from human
testing to
demonstrate
cross-contamination
safety.
Next slide,
please.
So you may not
be able to see
these pictures.
This is a first
generation MUNJI
device. I think
that those are
familiar with
these
technologies
know what that
device would be
called. And you
can see after
injection into
the test
fixture, there
is contamination
at the injection
site. There's a
combination of
splash back as
well as contact
contamination
during the
injection
process. You see
that it's
contaminated
with the
fluorescein dye.
The same is true
for the
protector cap
injector. This
is the protector
cap on the
nozzle face
itself. It's
hard to see in
this photo, but
this protector
cap post
injection into
the test fixture
is also
contaminated.
But the down
stream sample
collected after
injection into
the text fixture
is demonstrated
to be free of
cross-contamination.
Next slide.
So this is a
slide showing
the comparison
of first
generation MUNJI
testing with
this method
versus a
protector cap
injector. These
are the number
of samples. So
for a 100
samples with the
first generation
MUNJI device,
all were
contaminated, a
100 percent with
an average
contamination
rate of 268
picoliters. In
comparison with
the protector
cap injector for
300 samples, all
samples were
free of
cross-contamination.
So the
conclusion is
that the
protector cap
prevents
fluorescein
contamination of
the fluid path.
And, again, we
believe that
this is a very
useful and
powerful method
to demonstrate
contamination
risk with the
earlier devices
and then lack of
that risk with
the new
generation
protector
injector.
Next slide.
So for human
safety testing,
as I said, we've
been focusing on
detection of
hepatitis B
virus. And given
the
recommendations
from the WHO
Committee from
last year, we
focused on
recruiting high
titer
individuals that
have greater
than a million
copies per ml
and injecting
them with
buffered saline,
and then
collecting the
next dose and
assaying that
for presence for
Hep B and A.
Currently we're
implementing a
pilot study in
Pasadena,
California at
the Huntington
Medical Research
Institute, the
Liver Center
there. Working
with Dr. Myron
Tong.
We're focusing
on recruiting
high titer
volunteers, as I
said, but also
to HBV negative
volunteers as
controls. And
one key feature
of this study is
that it's a
nonsignificant
risk study by
our definition,
that the fluid
path is
sterilized
between use with
different
volunteers. And
so there's no
chance of
cross-contamination.
We're using an
assay that was
developed and
actually
licensed for use
in terms of
blood screening
products in the
United States by
National
Genetics
Institute. It's
called Ultraqual.
And it's also a
NAT assay. It's
a nucleic acid
test. So it's a
very sensitive
test. And I have
results from a
validation study
that was
conducted last
year prior to
initiating the
safety study
which was
started
September of
last year to
demonstrate the
sensitivity and
the limited
detection for
that particular
test.
This did receive
both PATH IRB as
well as
Huntington IRB
approval. And
we're currently
continuing to
recruit
volunteers for
this study.
Next slide,
please.
So, the study
endpoints
primarily is to
determine if
there's HBV
contamination in
down stream
doses. But
secondarily,
we're also
assessing the
pain of the
injection site
and any
injection site
reaction. So
that's also
collected in
terms of the
study, the
information from
volunteers.
As I mentioned,
there's two
sterile saline
injections per
subject, one in
each deltoid. So
after injection
into the
deltoid, the NET
is collected.
And then that's
sent off to NGI
for testing.
There's also
four negative
control samples
per volunteer
that are being
collected. Two
injector samples
prior to
injection into
the deltoid that
are collected to
demonstrate that
the injector is
free of
cross-contamination,
but also to
determine if
there is any
background
contamination of
HBV in the
examination room
where the
injections are
taking place.
Also two air
samples are
collected. These
are test tubes
that are left
open in the test
tube rack right
adjacent to
where the
injections are
taking place in
volunteers. Once
the injections
are completed,
then those are
stoppered and
the whole group
of samples are
sent to NGI.
Additionally,
another blood
sample is
collected the
day of
injections to
reconfirm titer
levels. So for
initial
enrollment there
is a blood test
that's conducted
to determine
titer level, and
that's a
condition for
enrollment into
the study. And
then the day of
injections
there's another
blood draw to
demonstrate that
there is still
high titer
viremia in the
particular
volunteer.
Next slide.
So in terms of
the assay
itself, this is
used for blood
product
screening in the
United States.
And it's also
uniquely used by
the Liver Center
for HBV titer
level
determinations.
It's part of
their clinical
diagnoses
screening. And
HMRI has a close
relationship
with NGI, and
that influenced
our decision to
work with both
HMR as well as
NGI.
It was validated
for use in the
pilot safety
study last July.
And the mean
sensitivity was
determined to be
1.589
internationally
in its per ml,
which is about
5.4 viral
copies.
The 95 percent
detection limit
is determined to
be 6.316
international
units, which is
equivalent of
21.73 copies. So
what it means in
terms of a half
cc volume, it's
about 10 viral
copies that is
reliably
detectable with
this method.
Next slide.
So this may be a
little hard to
see. To date we
have recruited
five volunteers.
I have to say
that it's been
challenging to
identify and
recruit and gain
consent from
volunteers.
You know, we've
talked about 10
to the 9th as an
average in terms
of viral load,
but in terms of
this Liver
Center and the
majority if not
all the patients
are hepatitis B
infected, it's
very difficult
to identify
those that are
greater than a
million copies
per ml. We have
identified
several that
have consented
to be in the
study; actually
three to date.
And as I said,
we're continuing
to enroll
subjects. We've
also recruited
our negative
volunteers. So
these volunteers
002, 004 and
005, those are
hepatitis B
infected
individuals. You
can see that
there's a range
of 10 to the
6th, 10 to the
8th in terms of
viral load. All
the down stream
samples from the
left and right
deltoids have
been negative
for presence of
hepatitis B DNA.
So we believe
that this is a
very powerful
method to
demonstrate
cross-contamination
safety with
human volunteers
focusing on the
infection of
interest,
hepatitis B
infection and
using a very
sensitive method
for that
detection.
Next slide.
So from that
pilot study our
plans are to
then proceed to
a larger scale
study that would
be conducted in
China. The
reason for that
is that in China
there is a very
high prevalence
of hepatitis B
infected
individuals,
more so than in
the United
States. There's
also a higher
prevalence of
higher titer
individuals. And
so we think that
it'll be much
easier to
recruit those
individuals and
then add to the
safety profile
for the
technology.
The current
study design is
focusing on
recruiting 300
high titer
volunteers. Each
volunteer would
receive two
injections. So
it would have a
similar design
to the pilot
study. We're
using the same
assay, and so
the jet injector
down stream
samples that are
generated in
China will be
shipped to NGI
for analysis.
The location
will be in the
Beijing area.
And there are
three sites,
three hospitals
that are focused
on hepatitis
treatment that
have agreed to
participate in
the study.
And we're
working with a
clinical
research
organization.
It's an MDS
Pharma office
based in Beijing
who help manage
and coordinate
the study
working together
with PATH and
Felton
International.
And I'd like to
close by saying
that the data
that's
generated, we
plan to submit
that in a
submission to a
national
regulatory
authority,
perhaps it's the
FDA, perhaps
it's the Chinese
SFDA. And we
were very
supportive of
the FDA's
efforts to
determine a
pathway to
demonstrate
safety of the
technology. And
we offer our
assistance to
help work with
your group to
determine a way
forward. And we
firmly believe
that there can
be a way forward
to demonstrate
that the
technology can
be safe.
With that, I'd
like to
introduce Dr.
Mark Kane, who
is my colleague
at PATH. And I
would say that
he is a
hepatitis B
expert. He would
like to make
some comments
regarding
earlier points
that were made
this morning.
Thank you.
DR. KANE: Thank
you.
These are more
observations of
things I've
heard today and
don't represent
in anyway any
kind of official
industry stance,
but just some
comments that I
had. I didn't
know where else
in the program
to be able to
insert them.
I think in 1984
by necessity,
because of the
level of
technology and
understanding,
the issue of
transmission was
defined as a
volume --
CHAIRMAN
EDMISTON: Excuse
me. You're not
on the list that
we had here. But
I appreciate
your being here.
But could you
make some
statement in
terms of
possible
conflict of
interest?
DR. KANE: Okay.
My name is Mark
Kane. I work at
PATH, so I have
exactly the same
conflict of
interest profile
as Dr. Zehrung.
Also worked for
20 years in the
hepatitis branch
of the Centers
for Disease
Control, the
last ten of
which at the
World Health
Organization.
CHAIRMAN
EDMISTON: Thank
you.
DR. KANE: Okay.
I'm sorry.
As I said in
1984 the issue
is framed as a
volume issue in
terms of
picoliters of
blood that may
or may not be
infectious, but
we're way beyond
that now in our
understanding of
how many genomes
and viral
particles might
be in a ejectate.
And so I think
it is possible
to ask questions
like given any
level of
detection in a
test system what
is the
probability that
there's one
infection dose
in that ejectate.
And it seems to
me that the
sensitivity and
specificity of
some of the
tests that we've
seen discussed
this morning
would make that
an answerable
question in the
real world.
The second issue
is that I
haven't heard
any reference to
the experience
with blood
screening using
the ELISA test,
which is
approved by FDA
for use in
screening all
blood. I
understand that
certainly there
are many
differences
between the
problem of
preventing post
transfusion in
hepatitis, but
there are also
are some
interesting
similarities.
And certainly
infusing an
entire unit of
blood versus the
volume of an
ejectate is
relevant, too.
And basically,
using an ELISA
which has a
sensitivity of
hundreds to
thousands of
picoliters
equivalents has
essentially
eliminated post
transfusion
hepatitis B in
the United
States. I think
the latest
estimate that
I've seen from
NAHs that
residual and we
may be getting
down into
compliance error
problems is
about 1
transmission for
220,000 blood
transfusion.
And so we have a
test of orders
of magnitude
less sensitivity
than the current
tests that are
available that
have essentially
done in a public
health sense a
very valid job
in reducing the
transmission of
disease.
The next point
has to do with
the model that
Martin
presented. And
when you present
a model, when
you multiple the
worst case
scenarios for
every variable
in your model,
in this case
probably ten,
and present that
as the results
of your
modeling, I
wonder whether a
better way of
presenting a
model is to take
your best case
estimates for
every variable,
multiple them
together,
present that as
the outcome of
your model. And
then you can use
the worst case
scenario and
even best case
scenario
estimates as a
sort of
confidence
interval.
Because it seems
that the greater
probability is
that the amount
of transmission
that would occur
using our best
knowledge of
what those
variables are
would be very,
very much lower
than the model
that was
presented to the
Committee by
Martin.
And the position
of WHO puzzled
me. In a sense
we were told
that there are
12 to 16 billion
injections given
int he world.
That 50 percent
of them were
estimated to be
unsafe.
Immunization
injections
account for less
than 5 percent
of those 16
billion
injections. And
they're pretty
much the only
ones that use AD
syringes. The
other 95 percent
of the 16
billion
injections
rarely use any
AD syringes, yet
the WHO position
is that they
cannot recommend
the device with
any theoretical
risk of
transmission
because if all
needle
injections were
given
compliantly,
there wouldn't
be any risk. To
me this seems
really a lot
like the perfect
being the enemy
of the good. And
I wonder if this
Committee would,
you know,
consider the
realism of that.
And that's
really all I
have to say.
I think that
there is a way
forward, given
our knowledge of
the infectivity
of hepatitis B
and the current
sensitivity and
specificity of
some of the
tests that had
been presented
this morning.
And I think that
as I imagine a
scenario and a
very bad weekend
when 300 million
Americans need
to be injected,
and doing that
with 300 million
single dose
vials of vaccine
with needles and
syringes seems
to me a very
unlikely
scenario. So I
think that there
is a potential
in this country
for a useful
high load jet
injector device.
And definitely
for mass
campaigns in the
developing
world.
So I would thank
the Committee
very much for
the opportunity
to address you.
Thank you.
CHAIRMAN
EDMISTON: Thank
you. At this
time I think
that there are
other public
speakers, other
speakers from
industry who
would like to
comment. But
because these
two individuals
that have
represented a
single entity,
I'd like to take
a few moments
now to ask the
Panel if they
would have any
specific
questions for
these two
gentlemen. Yes,
Dr. Butcher?
DR. BUTCHER:
Yes. I just
wanted to make
sure when the
presentation was
being made and I
understand we
didn't see the
video, but is
what happens
this is what you
passed to us,
this is taken
off each time
and is this
disposed or is
this sterilized
and then a new
one put on with
every injection?
Is that --
DR. ZEHRUNG:
Yes. Once the
injection is
provided into
the patient,
then the device
is reset. So the
device ejects
that spent
protector cap,
and then that's
discarded. And
then a new
sterile cap is
placed on the
nozzle face.
And you can see
in the
packaging,
actually the
packaging comes
in a tray of 25
protector caps
that can be
broken into rows
of five
protector caps.
And so I passed
some of the
pathing examples
around.
You would
present or open
one tray or one
protector cap at
a time, place it
on the nozzle
face of the
injector,
provide the
injection, eject
that spent
protector cap
and then open a
new sterile
protector cap
and place that
on the nozzle
face again and
proceed with
injections.
So for that, you
know the process
actually
explains why the
rate is lower
than the 900 an
hour or whatnot
that we've heard
with the earlier
MUNJI devices.
Does that answer
your question?
DR. BUTCHER:
Yes.
CHAIRMAN
EDMISTON: Dr.
Word?
DR. WORD: I
think I have two
questions, and I
think I just
need
clarification.
From an industry
perspective, and
I think you
partially
answered it,
when I looked at
where you've
done your mass
campaigns,
you've been
administering
things such as
meningococcal
vaccine, yellow
fever and polio.
Polio is
eradicated here.
We don't have
meningococcal,
we're not in the
meningococcal
belt. And we
don't have
yellow fever. So
essentially what
the message I'm
hearing right
now, because in
the beginning I
wasn't sure what
the question was
and it sounds as
if this isn't
something that
you want for use
in the United
States, but
you're looking
for use outside.
So then my
question really
is what is it
that you're
asking this
Panel to do?
Because why
wouldn't it be
for their
regulatory
agencies of that
specific
country?
You've alluded
to the fact that
oh we might go
to China. We're
interested in
hepatitis B,
etcetera. What
question do you
have, and I'm
not quite sure.
Are we here just
to provide
advice? Because
everything that
you've said I
don't see where
this is going to
be utilized
here.
DR. ZEHRUNG:
Right. Well,
actually, I
think that --
let me clarify
again. I work
for a nonprofit
organization, so
I'm not an
industry
representative
in terms of
being a
representative
for the
manufacturer.
Our focus is
health in a
developing
world. And so
when I listed
those mass
immunization
campaigns, those
are examples of
applications for
this technology,
at least as we
see it. But
there is also
the potential
application for
bioterrorism
response or
pandemic
response in the
United States.
That's something
that we are
interested in,
but as PATH as
an organization
that's not our
focus. That's
not the
constituents
that we focus
on.
But going back
to the FDA
meeting that was
held last year,
I think one of
the
recommendations
from that
Committee was
that WHO
deferred to
national
regulatory
authorities in
terms of
determining the
level of risk
and safety of
the technology.
So at least for
the United
States it's the
FDA.
For China, for
instance, and we
have already
initiated
discussions with
Chinese CDC and
a specific
program,
immunization
program
representatives
of China. We'll
work with their
NRA to license
this technology
in country
working with the
manufacturer. So
we will be doing
that individual
country NRA
submission and
interaction. But
at least for the
United States,
the question is
as the Panel has
laid out, is it
safe for the
United States,
what are the
methods that
could be used to
demonstrate
safety. And,
again, we defer
to the FDA and
the Panel to
determine that,
at least for the
United States.
The United
States is an
example around
the world in
terms of
national
regulatory
authorities. And
many regulatory
authorities
would follow the
FDA lead in
terms of
demonstrating
the safety of
the technology,
and there are
many examples of
that.
So I think there
is a connection,
although as you
say the focus
for the FDA is
consumer
protection in
the United
States.
So I hope that
answers--
DR. WORD: I
guess, too, in
all fairness for
the FDA is it
fair to have
them review and
do all the work
and not be
compensated for
it, for
something that's
going to be
utilized in
another country?
That's why I'm
saying it may
meet -- like
there are
standards that
will be set for
our government,
but it may not
be the same for
the others. And
that's why I'm
saying is it not
appropriate that
you go to their
regulatory
agencies and
find out what's
appropriate and
acceptable for
that particular
country? I mean,
it sounds like
you want them to
do the work and
not get
compensated.
DR. ZEHRUNG: No.
I think that
it's a
combination of
two approaches.
I think that
direct country
level
interaction,
which there's a
different risk
benefit profile
that they'll
evaluate versus
application of
this technology
in the United
States. So if
the FDA can
determine what
that risk
benefit profile
is and if it is
appropriate for
use in the
United States,
that is
information that
would feed into
a decision for a
local or like a
country level
NRA. Perhaps
they won't agree
with that.
Perhaps they
would agree with
a different risk
profile
calculation.
So it's a
combination of
the two
approaches, I
think.
CHAIRMAN
EDMISTON: Ms.
Petersen, do you
have any
questions?
MS. PETERSEN:
Yes, I had a
couple of
questions.
First, with
regard to the
information
about speed. The
presentation
notes that the
injector will do
six injections
per minute. Does
that include the
time to eject
the used cap, to
--
DR. ZEHRUNG:
Yes.
MS. PETERSEN: --
open another new
one to put it
on?
DR. ZEHRUNG:
Yes. We've
conducted time
studies in terms
of using the
device. And with
a proficient
user, someone
that's trained,
they can achieve
the six
injections a
minute rate. So
it includes
placing the
protector cap
on, filling of
the dose,
provide the
injection,
rejecting the
spent protector
cap, opening the
package.
MS. PETERSEN:
And will the
unit operate
without a cap?
DR. ZEHRUNG: The
current design
-- well,
actually the
design that was
cleared for
market last year
did not include
an interlock
feature. The
current design
that will be
actually used in
the China safety
study will have
an interlock.
And so that it
will require
placement of the
protector cap on
the nozzle face
for the device
to operate.
And it was part
of this sort of
the development
pathway that
felt, and then
perhaps Dr.
Loskutov could
speak to this,
first
demonstrating
safety in a
small pilot
study and then
having that
converge with
the design
development
effort to
include more
specific safety
features such as
this interlock
prior to actual
market
introduction.
And I should say
that these
technology is
not being sold.
It's not being
used in the
world. We're
actually working
with the
manufacturer to
build the safety
profile. And
we're looking
for NRA approval
and also public
health agency
approval for use
of the
technology. So
it's not really
being used. It's
not delivering
vaccines at the
current time.
We're conducting
safety testing.
Is that --
MS. PETERSEN:
And will the
newer version
with the
prevention
capabilities so
that a ap has to
be used, will
that be fairly
easy for an
individual to
modify so that
caps are not
necessary? I
guess what I'm
saying is can
someone buy it
and then get the
safety mechanism
off the gun so
they can use it
without caps?
DR. ZEHRUNG:
Well, I think
the focus of the
design effort
has been to
implement an
interlock
feature that's
very durable
that would be
very difficult
to disable. Now,
with tools or
whatnot, it
perhaps may be
possible. But
we've been
focusing on and
working with the
manufacturer to
develop a
feature that
would be as
durable and as
effective as
possible.
And if you'd
like to learn
more, we could
perhaps demo the
device for you.
But that's been
the focus. And
actually that
was a concern
earlier on
pre-WHO safety
meeting last
year working
with Dr. Bruce
Weniger and also
Dr. Mark Friede
at WHO that an
interlock
feature was an
absolute. If
this device was
going to be used
in the
developing world
for mass
immunization
campaigns, it
needed an
interlock. So
we've been
focusing on that
as an effort.
CHAIRMAN
EDMISTON: Dr.
Word?
MS. PETERSEN: Do
you have any
sense of the
cost of the
caps? Say the
device approved
and sent out for
use, what the
caps would cost?
DR. ZEHRUNG:
That's a good
question. The
spec has been
that the cost
per injection
needed to be
much less than
the cost of an
auto-disabled
needle and
syringe, at
least in the
developing
world. So the
cost of
auto-disabled
syringe is,
perhaps, down to
.04 cents, more
likely .05 to
.06 cents range.
It's been
projected that
the cost per
injection for
this device with
the disposable
protector cap
would perhaps be
.01 to .02
cents. So that's
another benefit
of the
technology; that
it's extremely
low cost. And
not only is it
needle-free, but
it's comparable
to -- well,
actually it
exceeds the
auto-disable
syringe costs,
but it's
comparable to
reusable syringe
costs. So that
was another spec
that we focused
on.
MS. PETERSEN:
And how does
that cost
compare with
prior MUNJIs,
the cost of use?
DR. ZEHRUNG:
Well, prior
MUNJIs did not
have a
disposable
component. There
were some
components that
needed to be
replaced in
terms of O rings
and whatnot, but
those devices --
and I can think
of an example of
like the Ped-O-Jet,
which was
perhaps $2,000;
the per
injection cost
amortized over
the life of the
device was very
low, fraction of
it essentially.
So it is more
expensive than
those earlier
devices, but the
reason for that
is that there is
a disposable
component, which
is a recurring
cost per
injection.
MS. PETERSEN:
Sure. But in
developing
countries would
not the
practitioners be
comparing the
cost of the
previous device
with the lower
cost to this new
one that has the
additional cost
associated with
the cap?
DR. ZEHRUNG:
That's a good
question.
Interestingly
enough, given
the stop of
using this
technology in
the developing
world, there's
been a turnover
with those
health care
workers, many of
which are not
familiar with
jet injectors.
Those health
care workers
have either
retired or
they've gone on
to different
parts of their
life. And so
when we've
interacted with
health care
workers in the
developing
world, you know
their first
question is
where is the
needle. So
that's part of
also our
challenge is
really
reeducating in
terms of the
benefits for
needle-free
injectors, be it
this device or
disposable
cartridge
injectors, as
Dr. Friede had
talked about.
And so there
isn't that
comparison.
Actually, the
health care
workers that
we've talked to,
and also program
managers, their
question is
safety. They
want to know
that it's a safe
technology.
They're
concerned about
speed, they're
concerned about
using the device
in terms of the
logistics of
cleaning and
sterilization.
They focus more
on that.
Many times it's
not those health
care workers or
program managers
that control the
money. It's
actually further
up the chain. So
there's a
difference
there. So there
isn't that
comparison
that's being
made.
CHAIRMAN
EDMISTON: Dr.
Layton?
DR. LAYTON: Yes,
I have several
questions. One
relates to the
bullet point
where you say
prevents
cross-contamination.
DR. ZEHRUNG:
Yes.
DR. LAYTON: And
can you say that
it prevents
splash back?
DR. ZEHRUNG:
That is prevents
splash back?
Splash back
occurs, the
protector
contains that
splash back and
then that's
discarded. So
splash back does
occur during in
the injection
into tissue, but
that protector
cap contains it.
So the splash
back does not
contact the
nozzle face and
then thus the
fluid path.
DR. LAYTON: So
it prevents
splash back to
the fluid path
way in the
nozzle?
DR. ZEHRUNG:
Right. So there
is a reusable
fluid path and
the nozzle
orifice that
generates the
high velocity
narrow injection
stream, that
stream passes
through the
protector cap
and into tissue.
So the injection
site splash back
or reflux is
contained within
the protector
cap.
DR. LAYTON: All
right. Next
question, is
this -- you've
only presented
information on
subcutaneous. Do
you have
anything on
intramuscular?
DR. ZEHRUNG:
That's a good
question.
Our focus has
been as a design
spec given the
prevalence of
subcu or
vaccines that
are delivered
subcu for mass
immunization
campaigns, we
would consider
testing with an
IM specific
nozzle if that
were requested.
But I do not
have data on IM
delivery.
DR. LAYTON: All
right. Thank
you.
The final
question is
either you or
the FDA,
possibly. You
said you have a
special 510(k)
and the label
says that it's
an IDE. Why? Why
was --
DR. ZEHRUNG:
It's IEE.
DR. LAYTON: Why,
if says an
investigational
device, limited
by U.S. And you
said it was a
special --
DR. ZEHRUNG: Oh,
the packaging
for the
protector cap.
These are
actually samples
that I passed
out. So I'd have
to defer to Dr.
Loskutov and
Felton
International in
terms of
describing their
current labeling
and instructions
for use. But I
just passed
those out as
samples.
DR. LAYTON:
There was a
510(k) --
DR. ZEHRUNG:
Yes.
DR. LAYTON: --
for this
protector cap.
DR. ZEHRUNG:
Actually, there
is an original
510(k) for a
device called
the BI-3M, which
is precursor to
this technology.
It utilized a
different
protector cap.
And actually,
the Russians
over the last 15
years had
identified this
as a needed and
started with
very crude
protector cap
designs, and
it's been
refined over the
years.
So that original
510(k) was the
basis for the
special 510(k)
that was cleared
last year.
DR. LAYTON: All
right.
DR. ZEHRUNG: For
this new device
iteration.
DR. LAYTON: All
right. Thank
you.
CHAIRMAN
EDMISTON: Any
other questions
from panel
members? Dr.
Word?
DR. WORD: Just a
question about
this cap here.
You said it
can't work
without it,
correct? What
happens if
someone just
forgets to take
it off and
change it
between the
patients? I
mean, is it
designed that it
only can work
one time?
DR. ZEHRUNG:
Yes. The
interlock
feature requires
that the user
follow all the
steps for
filling and
ejecting the
spent protector
cap. So you
could not use
the cap or use
the injector
again with a
spent protector
cap. It would
force the user
to eject that
spent cap.
CHAIRMAN
EDMISTON: Dr.
David?
MR. DAVID: Thank
you. I have a
couple of
questions.
One, is you
design
associated with
specific volume
that the cap is
protecting or
it's a general
statement you
have on your
product?
DR. ZEHRUNG:
Volume in terms
of splash back
or --
MR. DAVID: In
the injector?
DR. ZEHRUNG: Oh,
you mean the
fixed dose?
Again, that's a
spec that was
determined by
the volume
that's delivered
in mass
immunization
campaigns, which
is basically --
I mean that's a
fixed dose
that's used in
immunization
programs as well
as in
immunization
campaigns. So
other than
perhaps BCG, all
vaccines are
delivered a half
cc.
MR. DAVID: You
mentioned
sensitivity and
specificity. I
didn't
information
relating to
specificity.
DR. ZEHRUNG: Of
the Hep B DNA
tests or --
MR. DAVID: On
the fluorescein.
DR. ZEHRUNG: The
fluorescein
test? Well,
actually, it's a
very specific
test. And we
have a report
that we put
together that I
could provide to
you and the
other Panel
Committee
members that
goes into
greater detail
describing that
test.
MR. DAVID: My
last question
relating to user
skills. And I
think you've
described it as
logistics;
things that
involve with
cleaning,
sterilization
and maintenance
of the device.
The cleaning and
sterilization,
your study
included that
just as part of
a validating
study particle
or that's a
requirement?
DR. ZEHRUNG:
It's a
requirement. It
was a
requirement,
actually, from
the IRBs that
review the study
protocol. And we
conducted a test
to demonstrate
that the fluid
path can be
effectively
steam
sterilized.
Through a third
party laboratory
we conducted
bioburden
testing
introducing
bacterial
contamination
into the fluid
path, following
the cleaning
procedure in
this and then
also the
sterilization
procedure to
demonstrate that
the fluid path
post steam
sterilization is
sterile.
So not only is
it a product
requirement in
terms of
maintenance and
demonstrating
the device, the
fluid path can
be sterile, but
it was also an
IRB requirement
to allow for
approval of the
study.
MR. DAVID: And
how often would
you recommend to
do that?
DR. ZEHRUNG: It
really depends
on the usage,
and actually
that's another
design
development
effort that
we're
undertaking
determining what
the maintenance
life and cycle
life would be
for the
technology. I
think that
earlier devices
there was this
recommendation
for daily
sterilization.
We are
determining
study designs to
demonstrate if
after using a
particular
vaccine, such as
measles vaccine,
for several
hours would the
user have to
replace that
fluid path and
use a new
sterile fluid
path.
And actually I
didn't mention
this, but the
idea is that
with one
injector hand
piece and foot
pedal there
would be
multiple fluid
paths. So in a
centralized
facility, you
would sterilize
perhaps five,
six, half a
dozen or more
fluid paths and
then that would
be packed with
the injector and
then sent out
for injections
on site.
So if a fluid
path, either
there was a
malfunction or
if by our study
results we
demonstrate that
it needs to be
replaced more
than daily, then
the user would
then take the
old fluid path
off, put a new
sterile one on.
And those
sterile fluid
paths, the
intent is that
they would be
packaged within
a tyvek pouch,
so then they
would be sterile
to the point of
use. Once the
user is ready to
use that fluid
path, they would
open the pouch
up, install it
on the injector,
prime the fluid
path and then
proceed with
injections.
MR. DAVID: Your
mechanism, the
interlock that
you mentioned,
so do you have
any estimate on
a life cycle,
how many uses?
DR. ZEHRUNG:
Well, our target
in terms of
durability of
the device has
been a quarter
million
injections. We
have conducted
some initial
life cycle
testing, and
that will be
part of the
design
verification
work that will
occur with the
latest design
iteration to
verify that the
design does meet
that design
requirement
prior to any
introduction in
the marketplace.
MR. DAVID: Thank
you.
CHAIRMAN
EDMISTON: Any
further
questions by the
Panel?
Thank you very
much.
DR. ZEHRUNG:
Thank you.
I understand we
may have one or
two other
presentations
from industry
representatives.
Do we have any
further industry
representatives
in the audience?
Raise your hand,
please.
We have two?
Could one of you
come forward
first and
identify
yourself? And,
again, briefly
describe any
potential
conflict of
interest?
MS. D'ANTONIO:
Yes. My name is
Linda D'Antonio.
The name of my
company is DCI.
And in terms of
conflict of
interest we're a
needle-free jet
injector
manufacturer.
Not
manufacturer,
developer. We
are working on
disposable
cartridge
needle-free jet
injector.
And, actually, I
wasn't planning
to speak today
but I just
wanted to
address one
point. This
morning I think
it was during
Martin Friede's
presentation,
there was a
question that
came up about
high speed
devices and the
multiuse nozzle
jet injectors.
And I just
wanted to make
sure, to make
clear that there
are other high
speed
needle-free jet
injectors, those
being the
disposable
cartridge type,
which is the
type that we're
developing.
Our company is
developing a
high speed
disposable
cartridge jet
injectors for
mass
immunization
type use, for
use in the
military for
bioterrorism,
preparedness
kinds of things
response. And so
I didn't have
really more to
say than that,
other than just
to simply make
it clear to the
panel that there
are alternative
injection
systems to the
multiuse nozzle
jet injectors.
And I don't know
if my colleague
has more.
So if there are
any questions on
that, I would be
happy to answer
them. But just
my
clarification.
CHAIRMAN
EDMISTON: Thank
you.
Yes. Come
forward and
please identify
yourself. Again,
identify any
conflicts of
interest.
MS. CALLENDER:
I'm Kathleen
Callender from
Genesis Medical
Technologies.
And we developed
the Pharma-Jet
injector and our
disposable vial.
I am President
of the company,
so I do have an
economic
interest in it.
Our family owns
the majority of
the stock.
And it's kind of
been my mission
to do this. I've
been working on
it for probably
eight years.
Mine is
completely
disposable, and
it's a one time
use plastic
polypropylene
vial. So our
concept is to
prefill it and
to booster pack
it for ease in
use in third
world countries
as well as in
our grocery
stores and our
flu vaccine
clinics.
And we have been
marching through
the FDA, and got
a long way to
go. We're
cleared as a
Class II medical
device, but now
I understand I
have to go
through the
Office of
Combination
Products.
So I just also
wanted to let
you know that
there's some
other people out
there that are
trying to solve
the problem of
disease
transmission and
trying to get
rid of some of
the needles in
the world.
Thank you.
CHAIRMAN
EDMISTON: Thank
you.
Are there any
further comments
from industry?
If that's the
case, let's move
on.
I would now like
the FDA to
present the
questions to the
Panel. I'd like
the questions
presented in
total, and then
we'll go back
and discuss each
question
individually.
MR. LIPMAN: The
first question
is: Identify the
scientific
questions that
need to be
addressed to
demonstrate
whether MUNJI
devices are safe
for multiple
patient use in
the United
States.
Second, discuss
the adequacy and
feasibility of
the currently
available
methods to
assess the
potential for
cross-contamination
and the risk of
disease
transmission by
MUNJI devices.
And finally,
Feinman, et.al.
in 1984
suggested that a
volume of blood
as small as 10
picoliters can
transmit
hepatitis B
virus in
chimpanzees.
However, this
finding is based
on a single
animal study.
Considering the
potential public
health benefit
of MUNJIs is
there a
threshold of
volume of blood
contamination
that presents an
acceptable risk?
If so, what
threshold would
be considered
acceptable?
CHAIRMAN
EDMISTON: Okay.
Could we go back
to question
number one. And
let me comment
before I open
this to the
Panel that the
issue at hand is
really the issue
of safety and
cross-contamination.
We're going to
address the
issue of whether
or not these
devices are
safe. And if we
have an issue
regarding the
safety of these
devices in terms
of
cross-contamination,
then what the
technologies or
the tests that
must be applied
to validate
their efficacy?
Again, the first
question:
Identify the
scientific
questions that
need to be
addressed to
demonstrate
whether MUNJI
devices are safe
for multiple
patient use in
the United
States.
And this time
I'd like to open
this up to the
Committee, the
Panel for any
commentary. Yes,
sir, Dr.
Butcher?
MR. DAVID: Mr.
Chairman, the
thing that I
would like to
ask is that we
have a
definition of
the MUNJI
devices now.
We've been
presented with a
few hybrids or
alterations or
advances and all
like that. Is
all of that
going to come
under the MUNJI
device or we
just sticking
with MUNJI?
CHAIRMAN
EDMISTON: We're
focusing on
multiple use
devices.
MR. DAVID: Okay.
DR. ARDUINO:
Well, I might as
well start.
I think if you
look at most of
the studies,
we're looking at
a poor surrogate
of blood
contamination.
So instead of
focusing on
blood
contamination
with the testing
that is
available now,
supposedly
molecular
testing for DNA,
we should be
actually doing
more studies to
look at to see
if we actually
have virus carry
over in your
injections or
cross-contamination
that way.
Because I have
problems with
using serum
albumin. It's
just too much of
it could be
leaked to too
many false
positives. And
if you look at
some of the
studies, even
their negative
control -- you
know, some of
the negative
controls were
positive with
those as using
that. Or we have
to find some
other indicator
of blood
contamination
there.
So I think we
should be
looking at the
infectious
agent.
CHAIRMAN
EDMISTON: Any
further
comments?
DR. BUTCHER:
Well, again, my
comment if to
follow along
with what was
just said, is
that it seems as
though all of
the studies that
we listened to
were previous
studies and none
of them seemed
to be updated
and so forth. So
it looks as
though we're
going to really
need to have
some concurrent
studies as to
what's going on.
CHAIRMAN
EDMISTON: Dr.
Layton?
DR. LAYTON: Yes.
In terms of
scientific or
engineering
questions, I
think you
definitely have
to put some
definition in to
the three
different
intended uses.
And what I'm
saying that is
intramuscular or
intradermal or
subcutaneous may
all require
different volume
ranges,
different
pressure ranges.
And these are
going to play a
role on the
amount of splash
back and also
the amount of
potential
contamination.
So definition
needs to be
established
relative to what
those
performance
criteria are for
those three
different
applications
intended use.
CHAIRMAN
EDMISTON: Let me
ask this
question and
toss it out to
the Panel: Do
you think that
there is
sufficient risk
in the use of
these devices
that warrant
consideration of
whether or not
these are safe
devices as they
currently exist?
MS. PETERSEN: I
think it may be
that if we
provide
recommendations,
we want to
create
recommendations
that take into
account
different
scenarios under
which they might
be used in the
United States.
You know, we
keep hearing
about the
bioterrorism,
and if you had
to vaccinate
3,000 people or
300 million in a
weekend how
would you do
that? And that's
certainly one
scenario. And at
that point we
would be willing
to accept some
level of risk
that I suspect
is very, very
different for
wanting to
vaccinate the
1500 first
graders in some
given town. And
it's handy to do
it one day and
kind of have it
over with, but
that's not a
pressing need.
You know, if
Junior can't be
there Tuesday
afternoon
between 1:00 and
4:00, it's not
going to be a
problem if he's
there Friday
morning or next
Wednesday.
What would be
okay I think is
very different
in those two
scenarios. And
there's also the
question of how
the military
component fits
in as well.
Because,
presumably,
that's not quite
the same thing
as the
bioterrorism
scenario.
CHAIRMAN
EDMISTON: So
what I'm hearing
from the Panel
members based on
not only the
information that
was presented
but the
information that
wasn't
presented, is
that there is a
relative level
of risk of
cross-contamination
with these
devices. And
that either
through increase
in technology or
through possibly
considering
alternatives
from these
devices, this is
the direction we
should be moving
in. Is that
correct? Is that
a fair
assessment?
MS. PETERSEN:
Can we quantify
the risk
associated with
the various
types of MUNJIs
and compare that
to other
possible devices
and tie that
into how it can
be used?
CHAIRMAN
EDMISTON: Well,
I think you
brought up a
very good point.
And where I'm
uncomfortable is
that in looking
at how these
devices are
being used is to
assess the risk,
the true risk
associated with
the use of these
devices. And
while I've heard
some compelling
information
based on both
personal and
laboratory
experiences, I'm
not really sure
from the
epidemiologic
perspective. I
have a good
handle in terms
of what the true
risk is in the
use of these
multiple use
devices.
I would
personally like
to see some
additional data
developed
looking at the
epidemiologic
nature. And I
think that data
is available in
a retrospective
perspective to
determine what
the relative
risk is.
Now, that's a
sort of a
personal
perspective
working in the
area of hospital
infection
control. But
does the rest of
the panel sort
of have a
similar concern?
DR. ARDUINO:
Well, some of
the EPI study,
or there's
potential EPI
studies to
actually go --
if we look at,
say, the VA and
the data they
have with their
elevated
anti-HCV rates.
If we were able
to go back and
look back to see
okay, now what
were the
exposures, are
there other
compounding
factors
involved. And
actually do some
sort of
formalized study
that actually
then will put
okay, we have
these risks
associated with
these -- well,
is there a risk
with a certain
type of device
or is it just
the categories
itself or are
there other
compounders in
there?
CHAIRMAN
EDMISTON: The
reason I bring
that up is that
in every device
which is
approved by the
FDA is a package
insert. And that
package insert
has a practice
in terms of how
that device
should be used.
And every device
has potential
for being
abused.
And I think
we've had
commentary from
some members of
the audience has
suggest, this
should be fail
safe device. I
don't see that
occurring with
the current
pre-amended
devices that are
currently in the
market.
So I think what
I would like to
see personally
from my
perspective is
some sort of
epidemiologic
data to really
give me a sense
of what the true
risk is within
both U.S.
populations and
the populations
that are Dr.
Friede is
dealing with
from the World
Health
Organization.
Dr. Lin, is that
a reasonable
question to ask?
DR. LIN: Well,
that's your
call.
CHAIRMAN
EDMISTON: Well,
you have to do
the work, all
right.
The other issue,
this is sort of
-- Dr. Word?
DR. WORD: I'm
sorry. As you
started to begin
to break down
the various
scenarios, which
I think is very
important, I'm
still not quite
sure if we
utilize these
same -- you use
these MUNJIs in
adults versus
pediatrics, do I
take a 2,000
gram infant
versus a 50
kilogram adult,
I mean -- I
don't know if
you've looked at
them in that
population. I
assume you have.
But I would like
to see something
in terms of
pediatric versus
adults and break
it down. Because
most children
are immunized by
-- they receive
the majority of
their
immunizations in
the first two
years of life
when they're
receiving them.
So if you're
saying -- it
sounds as if
we're utilizing
in the United
States, it would
be during a
situation where
we would have to
have a rapid
mass campaign,
and that would
really be
limited to some
type of
bioterrorism
type of thing.
Because if we
had a pandemic
with influenza,
we wouldn't have
the flu vaccine
available. It
wouldn't be made
anyway, so you
couldn't
administer it.
No one could
make it that
quickly. So
you're looking
at something
different.
But I also like
your comment
about why not
test it not to
look about see
if you isolate
viruses. So I
would second
that and also
just looking at
the route that
it's
administered.
CHAIRMAN
EDMISTON: I
think your
comment would
become
appropriate as
we move down to
the second and
third question.
DR. WORD: Oh,
I'm sorry.
CHAIRMAN
EDMISTON: The
issue that you
raise is the
relative safety
of these
devices, these
multiple use
devices in
pediatrics
versus adult
population.
If there is an
issue of safety,
and we're
talking about
cross-contamination
now when we use
the word
"safety," then I
suspect what I
would want to
know is there
technology
available which
would allow you
to retrofit, for
instance, the
pre-amended
devices? I think
this is a
compelling
argument for
using multiple
use devices, but
again it doesn't
address the
pre-amended
component. And I
think that's an
issue we have to
lay on the table
because there
are probably
thousands, if
not hundreds of
thousands of
these devices
still out there.
So is it
possible to
develop
technology
either similar
to this or
parallel to this
that would make
these
pre-amended
devices safe?
What's the
thoughts on
that?
MR. DAVID: I
agree with you,
Mr. Chairman. I
also would like
to add to the
scientific and
Terry said
correctly the
engineering
question is also
looking at the
life cycle. If
we're talking
about high
pressure, high
flow devices it
will be
appropriate to
look at benefit
risk ratio when
the device is
used for 100
thousand
injection as
compared to the
first or the
second
injection. And
what is the
performance
effect of that?
And that
question does
not have an
answer today.
CHAIRMAN
EDMISTON: And
does the risk
decrease or
increase with
longevity of the
device?
MR. DAVID:
Right. Correct.
CHAIRMAN
EDMISTON: Are
there any other
comments
relative to that
first question?
Let me review
that again; is
there data
available on the
relative risk of
these devices
within both U.S.
and world
population? That
would be
important
information to
have from a
scientific
perspective.
Number two, what
data exists
looking at the
safety or the
potential for
cross-contamination
of these devices
between both the
pediatric and
the adult
patient
population?
Number three, if
there is indeed
a risk, what
technology is
available that
either is in
place on new
devices prior to
approval or in
devices that
could be
retrofitted to
the devices
already that
have been
approved through
pre-amendment?
And the fourth,
which is an
interesting
consideration,
is that as these
devices age is
there any data
to validate the
safety component
of these devices
as they move
through their
life expectancy
of 100,000 or
200,000
injections?
Dr. Lin, is the
FDA satisfied
with the
response for
that first
question?
DR. LIN: Well,
you want to put
me on the spot.
CHAIRMAN
EDMISTON: You're
sitting at the
table.
DR. LIN: Yes. I
think this is
probably is a
very course of
this and then
the
recommendation.
CHAIRMAN
EDMISTON: Having
gone through
this once
before, what
we've been
presented with
today at least
in my mind has
given me a
better concept
of how we can
crystalize some
of these answers
that we couldn't
do six years
ago.
DR. LIN: Yes.
But you looked
-- the bottom
line of our
question
essentially
that, for
example, today
or tomorrow a
manufacturer
present, such as
one we have, but
any new
generation
device come to
us, then you
remind when we
talk about fond
memories, what
type of an issue
we should ask
the manufacturer
to address other
than, you know,
we know that
this is some
potential,
whether it's
perceived or
it's real or a
close
combination. But
what type of
question we
should ask. I
think you point
out -- that's
probably beyond
what the
pre-market
review we can
do. But if
somebody come to
us, as I say,
either today or
tomorrow or in
the next few
months, what
type of
questions,
scientifical
question we
should ask in
view of those
potential
cross-contamination.
You can help
with, that would
be assuming --
CHAIRMAN
EDMISTON: Any
more comments on
that first
question? Let's
move on to the
second question.
DR. WORD: I know
you've asked
about
cross-contamination.
Are you asking
us to specify
what specific
agents that
we're looking
for?
DR. LIN: No. I
think it's
scientific
equation for
some part right
now.
DR. WORD: Okay.
DR. ARDUINO: Or
the type of
testing? Are you
kind of aimed
at, you know --
we know that
splash back is a
problem. What
type of testing
have you done to
show that the
device does not
get
contaminated.
DR. LIN: Right.
CHAIRMAN
EDMISTON: And
this is from the
manufacturer's
point of view.
DR. ARDUINO:
From the
manufacturers.
CHAIRMAN
EDMISTON:
Because they're
going to be
responsible for
conducting these
tests.
DR. ARDUINO:
Yes.
DR. LAYTON:
That's the
questions that
the FDA asks.
DR. WORD: Okay.
DR. LAYTON:
Because they're
asking those
questions
relative to
industry. And a
lot of them is
how much splash
back does your
new device have
and how does
that compare to
the predicate
device.
DR. ARDUINO: And
if there is
splash back,
what engineering
controls have
you included
your design to
prevent
contamination of
the fluid
pathway. Kind of
wind that up.
CHAIRMAN
EDMISTON: Yes.
That works well
for the new
devices coming.
DR. LIN: New
devices, yes.
CHAIRMAN
EDMISTON: But it
doesn't address
the
pre-amendment
devices. And I
suppose we could
take a position
right here that
these multiple
injection
devices are
totally unsafe
and they
shouldn't be
used at all. But
I haven't seen
the data that
compels me, at
least from my
perspective, to
agree with that.
I mean, how do
you feel about
this as a Panel?
MR. DAVID: I
feel that you
definitely
raised the
correct
question, and
that's not only
we looked at --
reassociated
with data that
was presented,
but data that
was not
presented
concerning. And
my feeling is
that definitely
we need to send
a message of
cautious --
CHAIRMAN
EDMISTON: It may
very well be
that based on
these initial
questions that
we proposed,
especially the
first question,
is that the risk
is significantly
high in selected
patient
populations. And
based on that,
then possibly
these multiple
use devices may
not be safe. But
I don't think we
have that
information at
hand to make the
decision, or
even to make
that
recommendation
to the FDA.
DR. LAYTON: No.
I agree. We
don't have. But
it's suspect.
DR. WORD: So
would you
consider any --
say if you
cultured, you
did a viral
culture or PCR,
whatever, and
you isolated any
virus, would
that be
considered
unacceptable?
Because if I --
you know, if
it's 1 in a
million cases
that it happens,
if you're that
one it becomes
important. But
then, too, I may
be willing to
take 1 in a
million, just
like with a lot
of vaccines that
have adverse
effects, you
have to do a
million people
in order to see
it to protect
the good. So
then if you
limit it back to
the scenario
that you would
utilize it in
the United
States, then you
might say, you
know this is
worth it.
CHAIRMAN
EDMISTON: You
make an
excellent point.
Because it
always comes
down to risk
versus benefit.
And it may very
well be the
vaccine itself
has greater risk
associated than
actually the
injection
component. So,
there's a lot of
data we don't
have here and
hopefully we can
develop some of
this data over
the next couple
of years.
DR. LIN: Well,
but you know we
probably cannot
wait for another
couple of years.
But, as I said,
if the device
come in, then
what -- for
example, present
some of their
test data. For
example, they
have a study or
have a full
test,
unfortunate that
data is not
available yet.
But now that the
question -- the
submission come
to us and not --
like today or
tomorrow, then
in your mind
that as our FDA
Advisory member,
what would you
advise the FDA,
what type of
scientifical
question would
you ask?
For example, as
Dr. Wood point
that maybe you
have limited to
certain patient
population or
those type of
questions.
That's what we
are looking for,
your
recommendation.
CHAIRMAN
EDMISTON: Yes?
MR. WATSON: I
just wanted you
to put yourself
in the
reviewer's
position. You're
sitting at a
desk, somebody
drops this on
your desk and
says, you know,
evaluate this.
What question
would you ask of
that
manufacturer.
And I think you
were going that
route. I think I
heard some of
the comments
were heading now
scientific
testing, that
kind of thing.
That's really
what we want to
know. Because
we're in the
situation where
we will see
more, probably.
And we want to
make sure we're
at least asking
the right
question,
realizing that
maybe we don't
have quite yet
the cut off
levels, if you
will. Maybe that
will come later.
But having some
appropriate
questions to be
asked will give
us a good
starting point.
And I think you
started down
that road. So I
just wanted to
encourage that.
CHAIRMAN
EDMISTON: Well,
we know splash
back occurs. We
know it occurs.
And we know that
there's a risk
associated with
that. We don't
know how
significant that
risk is. One
tack you might
take is that
devices that are
being submitted
have to have the
ability to
reduce the risk
of splash back.
And that would
be a reasonable
expectation
understanding
that splash back
is a risk. So
that fits into
that technology
component; what
technology is in
place or can be
placed, input in
place with that
device, these
devices that are
coming along to
reduce that risk
of splash back.
DR. LAYTON: But
it has to be
expanded to the
extent that the
splash back is
also over the
performance
characteristics
of the device.
The pressure
variation that
the device sees,
the shelf life
or the number of
uses. Also the
volume that it
injects.
So splash back
has to be looked
under all of
these
conditions.
splash back
leads to
contamination.
CHAIRMAN
EDMISTON: So in
devices
presented to the
FDA, whether
it's ID subcu or
intramuscular,
that there has
to be some
performance
criteria to
demonstrate that
there is a
reduction in
splash back?
DR. LAYTON: Yes.
MR. DAVID: I'm a
little bit
hesitant with
the word
"reduction."
Reduction from
1,000 to 999.
DR. WORD: You
have to define.
DR. LAYTON:
Reduction from
the predicate
device, for one
aspect of it. If
you have a
predicate device
to demonstrate,
to be able to
compare it to.
But the other
aspect is, you
know, just
having the
information on
splash back and
relating into
our next
questions that
we're addressing
provides a
tremendous
amount of
information for
them to help
make a decision.
CHAIRMAN
EDMISTON: Any
further
comments.
DR. WORD: I
guess I, too, am
a little
concerned about
that word
"reduction."
Because it's non
specific. I
mean, they could
down by one
percent, it
would be
reduced. So I
don't know how
-- I mean, quite
honestly maybe I
missed it or I
don't recall
anyone
quantifying how
much splash back
if you had. If
they quantify
how much splash
back that you're
getting now from
the one that you
have -- you
know, I don't
know if someone
picks an
arbitrary letter
-- I mean,
amount. I mean,
do you go down
by 15 percent,
do you go down
by 25 percent? I
mean, you're
setting a goal
for something.
Or something
realistic.
CHAIRMAN
EDMISTON: Well,
let me do this:
Let me bring Dr.
Friede, can you
stand up by the
podium. And I
could I bring in
the gentleman
from PATH, could
you stand up
next to him,
please?
Here's the
question. I'm
going to ask the
question from
the gentleman
from PATH. When
you designed the
system what was
the percent
reduction in
splash back
within your
system?
DR. ZEHRUNG: The
goal was
complete
elimination of
splash back in
terms of the
contamination of
the nozzle and
the fluid path.
In comparison to
predicate
devices such as
the Ped-O-Jet
device and the
earlier design.
Like, for
instance, the
fluorescein
test, we used a
threshold of 10
picoliters,
anything below
10 picoliters
was considered
not
contaminated,
anything above
was
contaminated.
And the tests
that we
conducted in
comparison with
the predicate
device was to
demonstrate that
at that
definition of
contamination,
the device was
free of
contamination.
CHAIRMAN
EDMISTON: So you
achieved greater
than 95 percent
reduction?
DR. ZEHRUNG:
Yes. Yes.
CHAIRMAN
EDMISTON: Dr.
Friede, let me
ask you a
question. With
that expectation
what's your
thoughts on a
multiple use
device or a
criteria for the
development of a
new multiple use
device that
would assume a
95 percent
reduction in
splash back, be
it IM, ID,
substitute,
subcu?
DR. FRIEDE: I
think seeing the
fluorescein
data, this is
the first time
that we have
seen a test that
appears to be
repeatable,
reliable, to the
extent that this
does not require
very, very
complex
technology that
can only be
performed in one
laboratory on
earth.
So as a
benchmark I
would say this
is beginning to
look like a very
good benchmark.
The only concern
that I would
have as a
scientist, and
this a personal
view, is that we
do not yet know
how the in vitro
splash back is
comparable an in
vivo splash
back. But I
think this is
the first time
that we have
seen a test
which shows
really
significant
reduction.
Now to put
numbers onto it,
I don't know.
But when you
look at theirs,
it is zero
contamination
using the most
sensitive
measurement that
we have ever
seen. So this
appears to be a
very good
benchmark.
CHAIRMAN
EDMISTON: Well,
let's hold that
assay component
thought for a
moment when we
get to our next
questions. But
in terms of a
percent
reduction, this
Panel has been
asked to make
recommendations
to the FDA. Do
you feel that a
greater 95
percent
reduction in
splash back is a
reasonable
expectation
given the
current level of
technology
that's emerging
with these
devices?
DR. FRIEDE: The
data that was
presented by
Darin suggests a
100 percent
reduction in
splash back.
CHAIRMAN
EDMISTON: So
you're
recommending
that we suggest
to the FDA that
there should be
a 100 percent
reduction in
splash back with
this device?
DR. FRIEDE:
There is a test.
It is relevance
may be called
into question,
but there is a
test which
achieved 100
percent
reduction in
splash back.
I bring you back
to the statement
that was made,
everything must
be viewed in
risk benefit.
CHAIRMAN
EDMISTON: It may
very well be
that a 50
percent
reduction gives
you the risk
benefit ratio
that you need.
That's the issue
that's sort of
before us.
DR. FRIEDE: For
each different
scenario of
acceptable risk.
CHAIRMAN
EDMISTON: Yes.
DR. FRIEDE: When
we've heard it
is completely
different to be
giving little
Johnny his
measle shot or
to be giving the
whole population
an antiterrorism
shot.
CHAIRMAN
EDMISTON: So it
would seem to me
that in your
population, the
population that
the World Health
Organization is
dealing with,
there may be an
intrinsically
higher risk in
some of those
subset
populations
compared to what
we see in our
own population.
Is that correct?
DR. FRIEDE: I
would say so.
CHAIRMAN
EDMISTON:
Therefore, the
performance
characteristics,
because
obviously these
devices are
going to be
submitted to the
FDA and other
nations
throughout the
world will be
using these
devices, the
performance
characteristics
really should be
applicable to
not just the
populations
here, but
obviously the
populations
abroad, which
will be at a
higher risk
category?
DR. FRIEDE:
Exactly.
CHAIRMAN
EDMISTON: Dr.
Word?
DR. WORD: I
guess I may not
necessarily-- I
would suggest
that we are not
here to approve
or make
recommendations
for other
countries. I
think they have
their own
licensing
organizations.
And that we, I
think, as a
Panel for the
FDA, that we
should set the
standard for
what is
acceptable for
the use in the
United States.
And then we say
this is what we
will find
acceptable here.
If you choose a
different
scenario outside
in another
country, then
you go to --
you're going to
go to their
licensing
agency. I think
that's fair. But
I think if we
try to break it
down for every
single -- well,
not every single
country, but for
different
regions of the
world, I mean
you could have a
PI that's so
long or even
just when they
submit an
application,
it'd be so long.
Plus, I go back
to I don't know
if necessarily
fair that U.S.
reviewers have
to do it for the
rest of the
world and not
get compensated
for it.
MS. PETERSEN:
Perhaps one way
to address that
issue as well as
to look at the
issue of risk
and relative
risk and what's
acceptable when
is for the FDA
to look very
seriously at
tying the use
and restricting
use to specific
scenario and
saying for this
purpose we'll
look at this
way.
One concern that
I have is that
we go forward
with this
because of the
issues of
injection in
other countries
and the genuine
need in other
places to have
such a system
and our concern
about
bioterrorism,
the device gets
approved and
then suddenly
it's being used
in ways where
that risk is not
really
appropriate for
the situation.
Mass vaccination
of school
children, for
example, or
pneumonia
vaccine for
older people who
may already be
somewhat
immunosuppressed
or have other
risk factors.
Where what we
think is, ah,
kind of, sort
of, usually
fairly
negligible for
most of us is
really much more
significant of a
risk. You know,
to tighten those
approvals for
use in
particular
scenarios and
rule out other
uses so that we
don't see the
drift of risk
into places
where it's not
really
appropriate.
CHAIRMAN
EDMISTON: You
know, in
scenarios such
as this is very,
very difficult
to define a
relative risk.
And I'm really
uncomfortable
from my own
perspective to
recommend a
percent
reduction, not
having all the
information at
hand. I really
believe we need
to have a more
sound
epidemiologic
model that tells
us what the
relative risk is
going to be for
these devices,
and then base --
base the
performance
criteria of new
devices coming
forward, again,
on that risk
within those
patient
populations.
MR. DAVID: Mr.
Chairman, I
agree with your
comment.
However, it
seems like that
we are presented
with
technological
options that we
suggest that we
can at least as
a benchmark
achieve a
tremendous
reduction into
the 90, even 200
percent of the
contamination
due to splash
back. So as a
Panel member, I
would like to
recommend that
we will ask the
FDA to achieve
this type of
benchmarking in
their
consideration of
the product.
CHAIRMAN
EDMISTON: In
terms of that
assay, has that
assay been
repeated by
other
investigators or
is that a single
observation from
your group?
DR. ZEHRUNG: It
has not been
repeated. It's
only been
in-house work at
PATH over the
last several
years.
CHAIRMAN
EDMISTON: You
know, this
really fits more
into the assay
component. But I
think we really
need to validate
that assay. And
I think the FDA
needs to
validate that
assay either
from other
independent
investigators or
in-house
contractual
sources.
But again,
relating to the
first question,
are there any
other issues
here within this
first question
that need to be
-- yes, sir?
DR. LIN: If you
allow me, can I
ask PATH
presenter a
question about
--
CHAIRMAN
EDMISTON: Yes.
DR. LIN: Do you
mind that I ask
you question?
DR. ZEHRUNG: No,
sir.
DR. LIN: And in
your -- I did
not have a
chance to hear
that your
product called,
but I have a
question. The
fluorescein dye
that you use is
water-soluble or
is it viscous?
DR. ZEHRUNG:
It's
water-soluble,
yes.
DR. LIN: It's
water-soluble.
But how much of
that would meet
actual blood
condition, you
know the blood
is kind of
viscous. And did
you see any
difference, have
you --
DR. ZEHRUNG:
That's a good
question, and it
goes to this
issue of in
vitro and sort
of replicating
the tissue
response. The
resulting dye
concentration is
very viscous,
but in terms of
comparison to
blood, I think
that's an
interesting
point to pursue.
We've focused on
the test as a
means to induce
contamination of
the fluid path
and using a
marker that's
actually very
inexpensive and
very innocuous
in terms of
safety, and that
could be easily
detected. But in
terms of the
protocol and the
reports that we
put together,
we'd be more
than willing to
share with you.
DR. LIN: Okay.
Thank you.
CHAIRMAN
EDMISTON: I
think in terms
of this
reduction issue,
because my
colleagues
really brought
to my attention
that reduction
is not a very
finite
terminology, but
one of the other
considerations
is possibly a
significant
reduction. A
significant
reduction in
splash back
compared to
predicated
devices. And I
think that would
address some of
the safety
aspects of this
device.
I want to thank
Dr. David for
bringing that to
my attention.
Yes, Dr. Friede?
DR. FRIEDE:
Could I just
make a comment
on that? Imagine
we have two
devices out
there. And take
the example of
the device that
was presented
where we have
absolutely no
contamination of
the fluorescein,
not whatsoever.
And then we have
another device
that's called X.
And this has a
contamination of
-- it's
significantly
better than it
was 30 years
ago, but we are
seeing that 1
out of every 10
shots is getting
contaminated,
and it is
getting
contaminated
with, let's say,
20 picoliters of
what would be
liquid. So let's
say 20
picoliters of
blood.
So would you
really consider
allowing that
device to be
used when there
is a safer
device
available?
From my point of
view and public
health sector, I
would be giving
recommendations
to using the
device that has
the safer
profile, even if
it was only
safety in terms
of theoretical
safety.
MR. DAVID: If I
can jump in. As
a Panel member
I'm not
convinced that
there is 100
percent safe
device. This is
a short
description of a
slide that has
not been
validated. So I
would love to
believe that
this the
mainstream
rather the
extreme, but we
don't have data
to say this is
the benchmark.
So I support
your view and I
would definitely
look at the
possibility of
looking at 100
percent
reduction as the
answer to the
first scientific
engineering
question. But I
realize that we
do not have
sufficient data
today to ask for
that. And by
suggesting
significant
reduction with a
p value that is
small, that's
getting close to
that.
CHAIRMAN
EDMISTON: I
think there's an
underlying issue
here that I'm
uncomfortable to
address, and
I'll bring it up
again, is that
your point is
valid. And I
think that as
the technology
improves, we're
going to see
devices that
have a
significant
impact on
reducing splash
back. But what
the predicated
devices, the
devices that are
already in the
field? What can
we do about
those devices?
And I think
that's the
troubling
component.
Because as you
remember asked
Mr. Lin are we
talking about
guidance for new
devices or are
we also
considering
those devices
that are
currently in the
field.
And my question
that comes up is
that I'm not
convinced that
these devices
are totally
unsafe from a
cross-contamination
perspective. I
think that when
we look at the
relative risk,
and that's where
I have a
problem. If I
had really
compelling
numbers, and
I've seen some
data that
suggests there
might be a
safety issue.
But they're
limited studies;
animal studies.
And also
possibly with
the application
of this new
technology, we
may be able to
get better data
on whether or
not these
devices
represent a
significant risk
for
cross-contamination.
It would be
difficult for me
to say at this
time to the FDA,
though there are
probably people
in the audience
who would love
for us to say
this, that these
multiple use
devices are
unsafe and
should not be
used under any
circumstance.
You might be
happy for us to
say that, too.
But I'm just not
comfortable
personally
making that
comment or
unless my Panel
members feel
overwhelming
that this is the
case and these
devices aren't
unsafe.
DR. BUTCHER: Mr.
Chairman, what I
would say is
that the
standard should
be set and the
preexisting
devices should
be brought up to
that standard
for us to say
okay.
So I don't see
it as a
difference. I
agree with your
point of view.
But if we're
saying a
significant
reduction, that
means any
pre-device
should have a
significant
reduction also.
CHAIRMAN
EDMISTON: And it
may be entirely
possible to
retrofit these
devices if
that's the
vendor's wish.
DR. BUTCHER:
Yes.
CHAIRMAN
EDMISTON: And
the vendor
wishes not to do
that, then these
devices may
actually go
away.
Any other
comments? Okay.
I think we can
move on to
question two.
Discuss the
adequacy and
feasibility of
the current
available
methods to
assess the
potential for
cross-contamination
and risk of
disease
transmission by
MUNJI devices.
And I think
we've probably
come close to
answering that.
DR. ARDUINO:
Yes. And
basically this
is detecting the
viral agent in
ejectate from a
device following
its use on a
none posit. And
you can that
now. With the
NAT testing and
PCR testing
that's
available, we
could probably
do that. And
actually figure
how many copies.
CHAIRMAN
EDMISTON: Any
other comments?
Do you think
there should be
a biological and
a physical test
in parallel when
testing these
devices? For
instance, the
fluorescein
would not be a
biological test,
per se. Would
you consider
that a
biological test
or a chemical
test, correct? A
chemical/physical
test?
What does the
Panel feel? Does
the Panel feel
there should be
a higher
threshold here,
not just a
single test but
at least two
tests to
validate the
ability of these
devices to
reduce splash
back?
MS. PETERSEN:
Well, I think if
you could do a
physical or a
chemical type
test in a human
population, you
would certainly
be getting a
better picture
of what actually
happens in the
clinical setting
in the field.
And you're
looking at the
physiological
barrier that
we're dealing
with, human skin
as opposed to
calf skin, mice
which has some
different
mechanics and
physics
associated with
it.
CHAIRMAN
EDMISTON: I
understand this
a new
technology, this
assay. Do you
feel it'll be
possible to
correlate this
assay with
biological
assays?
DR. ZEHRUNG: The
fluorescein
test?
CHAIRMAN
EDMISTON: Yes.
DR. ZEHRUNG: I
would recommend
that it be a
combination of a
fluorescein or a
chemical test,
plus a human
test with a
mark. And that's
why we've
focused on
hepatitis B and
conducting
clinical trials
as such.
I think that
these questions
of correlating
the in vitro
tests to an in
vivo sort of
model will
always be there.
And regardless
of how thorough
you would be in
terms of trying
to model that,
it would just be
easier to
graduate right
to an in vivo
model, such as a
human being.
CHAIRMAN
EDMISTON: Well,
there's other
models out there
for other types
of devices in
which they look
at both bench
type data and
also like
clinical trial
type data. So I
think that's a
valid approach
if these devices
are going to
come forward and
we're going to
demonstrate
their efficacy
and safety, then
the use of a two
or more test to
validate their
safety and the
prevention of
cross-contamination
is probably
warranted.
MS. PETERSEN: I
mean, at least
in that
circumstance you
would have a
more balanced
picture of the
risk and you
could, I think,
more easily
consider that
risk benefit
equation that we
keep coming back
to from all
directions.
CHAIRMAN
EDMISTON: Dr.
Lin, I think
we've been
presented data
both on the
bench and in
animal studies
and some limited
clinical data
which suggests
that there are
assays out there
at varying
levels of
sensitivity. And
I would suggest
that the choice
be made to
choose the most
sensitive assay
to validate the
safety of these
devices.
Any comment on
that from Dr.
Layton?
DR. LAYTON:
Don't forget,
you're going to
have
multivariant
data also
relative to the
splash back.
Just splash back
alone without
your biological
or your human.
But your
laboratory your
bench top is
going to give
you a tremendous
amount of data
showing what
that is for the
level and degree
of splash back.
So that's a
major part of it
also.
CHAIRMAN
EDMISTON: From a
guidance
document
perspective in
terms of the
manufacturer
should the FDA
then recommend
that two or more
assays be used
to validate the
safety of these
devices?
DR. LAYTON: I
don't have a
problem with
that.
CHAIRMAN
EDMISTON: Dr.
Word?
DR. WORD: I
don't know
should we say
one has to in
vivo, one
because -- I
mean you're
talking about --
maybe not. You
were suggesting
like with the
fluorescein,
that was -- and
I don't know if
we necessarily
have to use
fluorescein.
They may be able
to use something
else, just to
see if they're
getting
something back
to pick that up,
but something
that's done in a
human. But I
think your point
was well taken.
You want to know
if there's
actually virus
isolated.
DR. ARDUINO:
Because if
there's no virus
isolated and
we're not
getting virus --
DR. WORD: It's a
moot point.
DR. ARDUINO:
Then it's a moot
point.
CHAIRMAN
EDMISTON: Dr.
Lin?
DR. LIN: I think
the easy road to
travel that we
have been faced
right now as
this morning,
now FDA has
present from our
own laboratory,
CBER and some
people present,
although there's
some potential--
potentially this
is some -- the
FDA can
recommend to the
manufacturer to
do some kind of
a testing to see
whether this
splash back or
any residential
blood remaining
after its
injection. But
the current
suggests no any
-- that can be
directly applied
to a MUNJI
device. And that
is one of the
problems we are
facing.
I know that Dr.
Friede from --
last year he
convened a panel
of expert to
address this
issue. I know
whether, Dr.
Friede, you care
to comment to
see those
methods could be
used with it to
be applied to a
MUNJI device.
DR. FRIEDE:
Could you remind
me what this
assay was I --
DR. LIN: I
thought that
last year you
convened a panel
of experts to
replace the --
DR. FRIEDE: Yes.
The meeting that
we had in March
last year, we
looked at the
albumin assay
and we decided
that this was
inappropriate
because to
actually measure
as a surrogate
of safety.
Because you get
albumin and dead
skin. And dead
skin has no
value. You get
albumin on the
hair.
So at that time
we thought that
the measurement
of hepatitis B
as an example of
a live virus,
which has been
suggested here,
appeared at the
time to be the
most appropriate
in vivo assay.
And my gut
feeling is that
that concept of
having both in
vivo with an in
vitro, the in
vitro we've been
able to get much
larger numbers
to give you more
confidence so
that the two
together -- but
I think there's
many ways to do
the in vitro as
well; enzymes,
fluorescein,
other things,
coloring agents.
DR. LIN: But in
your mind, if
you don't mind I
can ask.
CHAIRMAN
EDMISTON: Of
course.
DR. LIN: In your
mind it is still
currently
there's any test
method that can
be applied to
the MUNJI device
so that FDA
want, FDA
reviews some of
the summation
then we say well
this is the test
you should do to
hear that this
device is safe
for multiple
use?
DR. FRIEDE:
There is no
validated assay
yet. I think the
assays that we
heard about
today, if
validated when
we see the data,
I think we'll be
able to look at
the data and
assess whether
these are
applicable. It
looks promising.
DR. LIN: But
it's not ready
yet --
DR. FRIEDE: The
committee did
not see all the
data last year
that we saw
today. And all
the committee
recommended last
year was that
the evaluation
virus was more
relevant than
evaluating human
albumin.
CHAIRMAN
EDMISTON: Could
Dr. Daya come up
to the podium?
Dr. Friede,
could you stay
there. I just
like listening
to you.
One of the
issues is
feasibility. And
there will be a
burden placed
upon the vendor,
the manufacturer
if the FDA
requires
testing.
Lt me ask you a
question. You
described very,
very well the
technology that
you're familiar
with that you've
had the ability
of performing in
your laboratory.
Does that
represent
feasible
technology from
a manufacturing
test
perspective?
DR. RANAMUKHA:
Before that I
would like to
comment on, this
-- I laid out
all the methods
available. I
wasn't aware of
the fluorescein
method, but the
published
methods. Out of
these published
methods the best
we can get is
100 copies, 100
genome
equivalents per
milliliter of
blood. With
that, you know
there comes what
is our detection
limit we need.
We don't know
what the limit
is. So with
that, we cannot
say the method
in that case.
Because that's
the lowest we
can go down. So
that's the
question that we
are dealing
with.
CHAIRMAN
EDMISTON: This
is a difficult
question.
DR. RANAMUKHA:
Yes.
CHAIRMAN
EDMISTON:
Because there's
a similar
question that
comes up with
TSE, as you
realize.
DR. RANAMUKHA:
Yes.
CHAIRMAN
EDMISTON: You
know, what is
the level of
infectivity in
terms of the
prion.
DR. RANAMUKHA:
Yes.
CHAIRMAN
EDMISTON: And
that varies
widely. And I
think what you
described and
what's been
brought to my
attention that I
wasn't aware of
is that the
infectivity is
going to be
highly variable
depending on
where the
inoculation is
being given.
DR. RANAMUKHA:
Absolutely.
CHAIRMAN
EDMISTON: So I
think that to
place an
unrealistic
burden upon
industry to
perform at such
a high level
with probably a
very expensive
test at this
point, it may
not be prudent,
nor may it be
fair unless we
have some sound
statistical data
showing the
efficacy of this
procedure.
DR. RANAMUKHA:
Yes. In that
sense, actually,
and like I said,
there are two
methods. One is
the HBV-NAT
assays and the
second is the
Taqman assay.
Taqman use
broader range
and also it is
feasible because
it does not
involve a lot of
expensive
equipment. So it
is a feasible
assay and then
it can be used
under diagnostic
setting, I would
say.
CHAIRMAN
EDMISTON: My
understanding
with the FDA in
the past when
they've had a
situation such
as this, they
looked at
available
technology and
then the vendor,
the industry,
the
manufacturers
have the option
to submit data
involving one or
more of those
technologies,
correct?
DR. RANAMUKHA:
Right. And
that's correct.
CHAIRMAN
EDMISTON: All
right. I think
the other issue
that comes up,
and I think this
-- and let me
get that fellow
that PATH again.
You stand up
there. All three
of you guys
stand together,
all right. A
Kodak moment
here.
As a
methodology, is
that fluorescein
a feasible
methodology from
industry's
perspective if
it could be used
by a variety of
vendors?
DR. ZEHRUNG: I
would believe
so. And it's not
only feasible,
it's extremely
low cost. And
the comparison
in terms of the
assay that's
being for the FE
testing, it's
very expensive.
It's $185 a
sample to test.
So that would
actually
represent a
financial burden
to a
manufacturer.
And we've
adopted that or
we've accepted
that sort of
responsibility
in terms of our
collaboration
with the
manufacturer.
But it's an
expensive test.
CHAIRMAN
EDMISTON: But I
know there's no
gold standard.
But that
probably could
be perceived as
a possible gold
standard if you
looked at the
rest of the
methodologies
out there.
I think the
fluorescein
assay has great
promise, but the
concern I have
is that I've
only seen the
data presented
from your
institution. And
we'd need to see
more data with a
variety of
devices.
So I suspect
what I would
consider to
present to the
panel is that we
have methodology
available that
the FDA could
require the
vendor to submit
performance data
using a number
of those current
methodologies.
If the
fluorescein
assay appears a
successful
assay, then I
would also
encourage the
FDA to consider
that as one of
the surrogate
tests. But I
think at this
time I'm not
sure if we could
recommend that
assay because we
don't have the
kind of
laboratory
experience with
that as we do
with the other
methodologies.
Any comments
from the Panel
on this? You
guys agree or
disagree or --
MR. DAVID: I
agree, yes.
CHAIRMAN
EDMISTON: Okay.
So I believe in
terms of
question two,
the methodology
that should be
applied should
be feasible and
should be
adequate. And we
currently have
presented
published --
published
methodologies
which are
accepted by the
scientific
community to
detect
biological
particles in
samples, both
blood and other
body fluids. Is
that a
reasonable
consideration.
DR. LIN: I
wanted to ask
PATH, is your
method is going
to be published
in an open
literature?
DR. ZEHRUNG:
That's a good
question. We've
discussed that
and I think
we've just
focused on
supporting any
sort of
regulatory
submission for
the data. But I
think we would
be open to doing
that.
CHAIRMAN
EDMISTON: Would
you accept their
data from a
regulatory
perspective if
it's not peer
reviewed
methodology?
DR. LIN: Well,
as long as it's
scientifically
sound, then we
don't have a
problem whether
it's published
or not.
CHAIRMAN
EDMISTON: Okay.
DR. LIN: But my
question is that
to be aware to
other
manufacturing,
if the method is
published, and
then somebody
could use their
method to
compare --
CHAIRMAN
EDMISTON: Is
this a
proprietary
methodology?
DR. ZEHRUNG: No.
I mean, we
haven't designed
any sort of
patent, you
know,
applications for
it. So I think
our position
would be it
would be free to
industry to use.
CHAIRMAN
EDMISTON: So you
don't consider
it proprietary?
Does that fellow
back there
consider it
proprietary or
--
DR. ZEHRUNG:
From Felton? No.
CHAIRMAN
EDMISTON: The
gentleman who
was with you?
DR. ZEHRUNG: Dr.
Loskutov?
DR. LOSKUTOV:
No.
CHAIRMAN
EDMISTON: No?
Well, I think it
would be in the
best interest of
the industry as
a whole if we
had that data
available to us
in a published
form.
DR. ZEHRUNG: And
as I said
earlier, PATH is
more than
willing to
collaborate and
share this
information with
these
technologies.
CHAIRMAN
EDMISTON: Are
there any other
comments on
question two?
MS. PETERSEN:
Has PATH made
any effort to
seek some kind
of grant or
collaborative
funding to
assist with the
higher cost of
some of these
assays to get
the validation?
DR. ZEHRUNG:
Well, that's
part of the
funding from the
Bill and Melinda
Gates Foundation
is to conduct
the safety
testing for the
protector cap
injector. And so
that would be --
CHAIRMAN
EDMISTON: Dr.
Friede, do you
have any final
comment on this
issue here?
DR. FRIEDE: No.
As I said, this
is the best
we've seen.
CHAIRMAN
EDMISTON: Excuse
me. You
pronounce your
Friede?
DR. FRIEDE:
Friede.
CHAIRMAN
EDMISTON: Friede?
DR. FRIEDE: Yes.
CHAIRMAN
EDMISTON: All
right. I had
that totally
screwed up,
didn't I.
DR. FRIEDE: I
think that from
what I -- my
personal view is
that this has
set for the
moment a
benchmark. This
is the most
sensitive we've
seen. It is a
100 fold more
sensitive than
anything else
we've seen. And
I think from my
perspective
we're going to
try to go for as
safe as
possible.
CHAIRMAN
EDMISTON: We're
talking about
the fluorescein
assay?
DR. FRIEDE: The
fluorescein
assay.
CHAIRMAN
EDMISTON: But as
a scientist also
you would want
to see that be
submitted for
peer review?
DR. FRIEDE: I
would want to
see it permitted
for peer review.
I would also
somewhere along
the line like to
see somebody try
and if possible,
look at this and
say how does
this correlate
with real life
situation.
Because we are
talking about an
in vitro
situation. And
so if this is
possible
somewhere the
line. But I do
like the fact
that it can
probably be
repeated in many
laboratories,
easily done and
we can probably
even have these
kinds of things
standardized.
CHAIRMAN
EDMISTON: And I
think that's an
excellent point,
but I'm not sure
that's the
purview of the
FDA. I mean,
that's something
that's going to
probably
conducted
independently,
or at least
within their
laboratories if
they have an
interest in
this.
Any other
questions in
terms of -- any
comments?
DR. LIN: I tell,
you would echo
Dr. Friede --
CHAIRMAN
EDMISTON: Friede.
DR. LIN: His
comment. Can
correlate those
and test results
with their
ongoing current
study. That will
be wonderful. It
will be an
excellent
correlation for
that. So data
can be compared.
CHAIRMAN
EDMISTON: Any
further comments
on question two?
Did you
understand our
response in
question two,
that there
currently are
feasible and
adequate assays
available which
the
manufacturers
could use to
benchmark their
device.
DR. LIN: Okay.
CHAIRMAN
EDMISTON: Number
three, Feinman,
et.al. suggested
a volume of
blood as small
as 10 picoliters
can transmit
hepatitis B
virus in
chimpanzees.
However, this
finding is based
on a single
animal study.
Considering the
potential public
health benefit
of MUNJIs is
there a
threshold volume
of blood
contamination
that presents an
acceptable risk?
If so, what
threshold would
be considered
acceptable? Any
comments by the
Panel?
DR. ARDUINO: I
think we're
focused on the
wrong thing
because it all
depends on what
the viral load
is on the person
-- you know. I
mean, this is a
single animal. I
mean, it's not a
population. I
mean, we have no
idea what real
infectious dose
is based on
what, an N of
what? And it's
kind of out of
context. Because
this wasn't in
vaccine
development.
This was more
looking at a
test development
for detection of
virus.
So, you know,
I'm kind of --
you know, 10
picoliters, how
we going to
measure 10
picoliters? It's
also based our
serum albumin
studies which is
probably a lousy
test to begin
with. So, you
know, how
accurate is that
really when you
look at the
other assays
they're using to
measure that?
I would still
look at
reduction of the
infectious agent
or you can
demonstrate that
there's no
infectious agent
or to whatever
the limit of
detection of the
tests are.
CHAIRMAN
EDMISTON: From
biological
perspective?
DR. ARDUINO:
From a
biological
perspective.
CHAIRMAN
EDMISTON: How
about the
concept of a
genome
equivalent?
DR. ARDUINO:
Well --
CHAIRMAN
EDMISTON: That's
the same thing?
DR. ARDUINO:
That's a copy.
CHAIRMAN
EDMISTON: Okay.
DR. WORD: I'm
sorry.
CHAIRMAN
EDMISTON: Dr.
Word?
DR. WORD: I
would probably
go with the
lowest level
detectable.
Because
generally if
it's not
detectable, we
say it's
negative if
you're looking
at copies of
something.
I'm not
comfortable with
that 10
picoliters at
all.
DR. ARDUINO: And
we're using
hepatitis B as a
marker, you know
it's great --
transmission
requires -- it's
great -- you
know, has more
potential
transmission
than the other
viruses.
CHAIRMAN
EDMISTON: Dr.
David, any
comment?
MR. DAVID: I
thought I have
an idea until I
heard the
comments. Now
I'm not
concerned. It
sounds to me
like you're
saying 10
picoliter is a
number you're
not comfortable
for several
reasons loading
N on one --
DR. ARDUINO:
It's all based
back on the
virus. You know,
you really don't
know what the --
and the viral
load depending
on, you know,
whether you're
HBV, HIV
infected, HBV
and HCV infected
or HBV alone, or
HBV with e
antigen. It's
going to be
different.
So I would
rather focus on,
you know, the
lowest
detectable
amounts of
virus.
CHAIRMAN
EDMISTON: Dr.
Word?
DR. WORD: I
guess my other
question is do
we have to
include other
viruses that
we've known are
blood born, not
just -- I mean,
I know hepatitis
B is the most
easily
transmitted. But
just like when
we screen for
blood, we screen
across the board
for hepatitis A,
B and C and
we'll also look
at HIV. Do we
have to set a
limit for all of
those, not just
hepatitis B? I
think I'd feel
more
comfortable,
because why not
treat it the
same way if
you're telling
me there may be
some back splash
in their blood
there. I don't
know how other
people feel
about it,
though.
CHAIRMAN
EDMISTON: Well,
we've been shown
methodologies
that exist that
detect anywhere
from one to 0.4
picoliters of
fluid
contamination.
You don't feel
that going down
to the -- one to
0.4 picoliters
would be a
sufficient
threshold?
DR. ARDUINO: No,
it's hard.
CHAIRMAN
EDMISTON: Dr.
Layton?
DR. LAYTON:
Well, I think
from an industry
perspective you
want to try and
put a limit on
it. And it goes
back and relates
there to the
question one
also. Now
whether it's 10
picoliters or
what is it, 0.4
or .4 picoliters.
And we're
looking at total
reduction of
splash back. And
you put a limit
on it today, you
don't know, you
know next year
we may have
another blood
born disease
that plays an
issue relative
to it. So that's
why the best
from an industry
standpoint and
from my
recommendation,
you try to put a
limit on it
relative to a
volume.
CHAIRMAN
EDMISTON: What
is the FDA's
perspective on
threshold.
DR. LIN: Well,
that's the
question that we
try to ask of
our panels to
help us. You
look at this
morning's
presentation,
this is
potential public
health need that
somebody
probably already
present. And
whether I say in
this country or
in other country
or in other
third world
country, this is
a potential
public health
need. On the
other hand, this
also has
potential risk
of blood
cross-contaminations.
Now if from
looking at the
risk benefits
consideration,
our question to
you is that in
your mind we
should not allow
any risk or
threats of risk
or we should
allow for
certain level of
risk. That's the
question that if
you can help to
address that, it
would be very
helpful.
CHAIRMAN
EDMISTON: Well,
what you're
saying is that
we get back to
that 100 percent
issue of
reduction in
splash back?
DR. LIN: Right.
Right. Yes.
CHAIRMAN
EDMISTON: And I
think if that's
the case, then
we can take it
one step
further, that's
no detectable
viral particle
or detectable
blood --
DR. ARDUINO:
Except we really
don't have,
other the
fluorescein,
which is a
surrogate, we
really have no
way of actually
measuring that
level of blood.
DR. LIN: I think
that that's why
you have to look
at question
number two,
which is also
the current
available
method.
CHAIRMAN
EDMISTON: Right.
DR. LIN: Is
there any way
you can really
direct to the
level. And if
not, then what
kind of level
you can
recommend to FDA
that would be
appropriate for
the reviews of
this type of
device.
CHAIRMAN
EDMISTON: Well,
if you review
the published
assays,
especially HBV
assays,
hepatitis B
virus assays,
you're limit of
detection varies
anywhere from .1
nanograms to 10
to the 9th,
depending again
on the
methodology. And
if you go 10 to
the 9th, we're
talking about a
threshold that
we initially
discussed early
on, anywhere
between 1 and 10
picoliters,
correct?
So we really
need some
consensus here
in terms of what
the Panel
believes to be a
threshold value.
And I think Dr.
Friede suggested
that 10
picoliters was
much too high,
correct?
DR. FRIEDE: I
think just
looking at the
numbers, we know
that 10
picoliters can
transmit
infection and
less than 10
picoliters will
therefore be
able to on some
occasions.
CHAIRMAN
EDMISTON: So on
a risk benefit
basis more than
likely what
we're talking
about is a
device that has
the capability
of reducing --
reducing
exposure to
below 10
microliters --
10 picoliters?
Correct?
DR. WORD: Excuse
me, Dr. Edmiston.
What is the
acceptable level
that we use in
blood when we
transfuse
someone? I mean,
I don't know
what the exact
number is. I
don't know if
anyone here
might know?
CHAIRMAN
EDMISTON: Dr.
Michaud?
DR. LIN: She's a
hematologist, so
she'll be --
DR. MICHAUD:
Ginette Michaud,
Deputy Director
of DAGID.
I would suggest
in fact the
question may not
be entirely
relevant to this
discussion.
Because I
believe that the
acceptable
limits are
driven by the
available
technologies.
And the tests
are applied to
blood products
which are
lifesaving
biological
products. So
it's really, I
think it's a
very different
thought process
that goes into
that.
And as you look
at the history
of screening of
blood products,
the limit of
detection on the
acceptable
assays has been
driven down as
the technology
is able to offer
a lower limit of
detection.
CHAIRMAN
EDMISTON: So
we're actually
back to the risk
benefit
component again
in the sense
that the
technologies
that we have in
front of us here
have variable
specificities or
variable units
of detection.
And I think that
Dr. Friede's
comment is that
if we're going
to try and
achieve maximum
reduction,
maximum risk
with overall
benefit of the
device, then we
need to look at
that value less
than 10
picoliters.
Would you agree
with that?
DR. BUTCHER: Mr.
Chairman, I
don't think that
we're going to
be able to put a
number on it,
but I think that
going to your
previous thing
that
significantly
reduce it down
and that would
get it. I mean,
we know that the
ten is too high.
We don't have
enough evidence
with those that
are less yet.
CHAIRMAN
EDMISTON: Yes,
sir?
MR. WATSON: I'd
like to put a
little
perspective on
the numbers. I
know that it's a
little bit of a
challenge to
actually pin it
down.
When we look at
substantial
equivalents,
which right now
what we're
actually
proposing to do
is set an
actually kind of
a bar, which is
a little bit of
a challenge in
the 510(k)
process, but it
can be done when
we have safety
concerns. If we
can get a number
-- realizing
that that is a
challenge, if we
can get some
kind of a bar to
start with.
Because we can
always make that
bar higher as we
know more about
the developing
technologies.
Right now we
don't -- I mean
10 picoliters
has come up or
10 picoliters
has come up and
we don't really
know what to do
with that.
CHAIRMAN
EDMISTON: Right.
MR. WATSON: As
you mentioned,
you know, less
than 10
picoliters, at
least if the
Panel -- and I'm
not suggesting
that they should
-- but if the
Panel would give
us that as a
starting point,
that would help
us tremendously.
But other than
that, it's sort
of a touchy
situation for us
because then we
don't have a
goal in place
without that.
So I would
encourage any
valuable numbers
that you think
based on what
you've seen
today, if you
can give us some
guidance in that
area, that would
be much
appreciated.
CHAIRMAN
EDMISTON: The
NAT technology,
can the NAT
technology
detect down to
10 picoliters?
NAT? I'm not
sure it can.
DR. RANAMUKHA:
NAT technology
detected DNA
copy numbers. So
it does not go
with the volume.
And so it comes
down to how many
copies you find
in the --
CHAIRMAN
EDMISTON:
Gotcha. Gotcha.
DR. ARDUINO:
That's why I
almost saying
that ten is
irrelevant if
you're looking
at how many
genomic
equivalence are
present.
DR. WORD: I was
going to say I
like presence --
CHAIRMAN
EDMISTON: Well,
the issue is is
that copies or
genomic
equivalents is a
nice ideal
number. But the
issue is also in
terms of volume
of detection.
And I'm a bit
perplexed by
this number
issue, you know.
MR. DAVID: And
the number is
what I'm left
today after
seeing the data,
is that 10
picoliter is
transmittable
volume and would
give a
significant
concern if we
are supposed to
judge risk to
benefit as the
safety issue is
not addressed.
So definitely
I'm looking for
a volume that is
below that
numbers, because
then I showing
this number is
unsafe.
CHAIRMAN
EDMISTON: And
realizing, too,
that number, 10
picoliters, was
based on a
single study,
correct? A
single study.
DR. ARDUINO: In
one animal.
CHAIRMAN
EDMISTON: In one
animal. But it
does represent a
starting point.
Does the
Committee have
any concerns
with making a
recommendation
of less than 10
picoliters as a
bar?
DR. ARDUINO: As
a start.
CHAIRMAN
EDMISTON: Start.
DR. ARDUINO: No.
DR. WORD: Can
you put the
caveat that they
will revisit it?
CHAIRMAN
EDMISTON: Oh,
they'll revisit
it, there's no
doubt about
that.
DR. WORD: No.
But if you tell
them.
CHAIRMAN
EDMISTON: This
is a moving
target. Am I
correct in
understanding
that this is a
moving target,
correct?
DR. LIN: Well,
when you set the
benchmark, then
I'm sure that
industry can --
but once you set
the benchmark,
the industry
would develop a
tendency to meet
that goal.
Hopefully.
CHAIRMAN
EDMISTON: You
represent sort
of the pragmatic
perspective
here, Dr. Friede.
You're out there
in the field. If
industry met
that benchmark
of less than 10
picoliters,
would that give
you a sense of
assurance that
we're moving in
the right
direction,
especially if we
evaluate the
risk versus
benefit
component.
DR. FRIEDE:
Okay. Let's just
imagine the
situation. We
have a device,
again this
famous device X,
and it's
actually
transmitting 5
picoliters per
injection.
That's less than
10. And we know
that we're in a
room with 50
percent chronic
carriers, and
they're all
looking very
yellow. And we
all have to
stand up and we
have to inject
the person
standing next to
us and then
inject
ourselves. How
many people here
are going to do
that, especially
if there was
another device
that was
undetectable
using the most
sensitive assay,
absolutely
undetectable?
So for me
putting a number
and saying five
or less than
ten, this is not
relevant. It
must be
undetectable
using the most
sensitive assays
that we have.
Because if you
can detect blood
on it, then
there is a risk.
That risk might
be, as Dr. Kane
mentioned, you
know I presented
the worst case
scenario and we
should also be
looking at the
best case
scenario. But we
are going to
have a
recommendation
to people to use
a device. And if
we know that
that device, we
say oh yes, use
it, it does
transmit blood
but not a lot.
Don't worry
about it. I'm
not comfortable
with that. I
think we have to
say there is no
detectable --
CHAIRMAN
EDMISTON: So in
essence we would
raise the bar to
a level we don't
even raise with
TSE?
DR. FRIEDE: Yes.
I don't know
what you do for
TSE, but it does
appear to me
that we have
seen this
morning data
which suggests
that there is
something that
does not have
any
transmission. It
appears to me
that this is a
benchmark and
one could not
allow anything
which is worse
than that.
CHAIRMAN
EDMISTON: What's
the Committee's
perspective on
Dr. Friede's
comment that no
detectable --
let me get a
vote here. How
many feel that
the Committee's
recommendation
for this
question should
be that there is
no detectable
entity?
Everyone who
feels there
should be no
detectable
entity. One,
two, three,
four. Four to
two or four to
three. So it
would appear
that the Panel
would recommend
that there would
be no detectable
entity.
I think that
that's a very
difficult thing
to achieve, but
that's the
Panel's
recommendation.
Any comments
from the FDA in
terms of that
recommendation?
DR. LIN: Well,
we can live
with.
CHAIRMAN
EDMISTON: You
can deal with
it?
DR. LIN: We can,
yes.
CHAIRMAN
EDMISTON: Okay.
All right. What
we're going to
do at this time
is take a brief
break or do you
want to continue
on. Well, I'll
tell you what,
let's have a
very quick
break. Let's
have a five to
ten minute break
and we'll come
back, and at
that time we'll
finish up with a
final public
comments.
(Whereupon, at
2:17 p.m. a
recess until
2:29 p.m.)
CHAIRMAN
EDMISTON: I'd
like to call
this meeting
back to order.
We will now hold
our second half
hour open public
hearing. If
there any
individuals
wishing to
address the
Panel, please
raise your hand,
identify
yourself at the
time. Also at
the time you
identify
yourself, please
indicate again
any proprietary
interests or
conflicts of
interest.
Please.
DR. KANE: Yes,
my name is Mark
Kane. I work for
PATH. I spoke
before and I
have the same
conflict of
interest profile
as the other
PATH speaker.
I think the
point I'd like
to make is that
is a perspective
that comes from
a little bit of
the history of
the development
of some of these
assays.
The original
intent in
developing the
serum albumin
assay, at least
at a certain
point, was that
we would in
parallel develop
a physical
chemical test
like the serum
albumin assay in
parallel with a
biological test
looking at the
actual etiologic
agent that we
were most
concerned about,
which is
hepatitis B.
Then because
although the
hepatitis B
testing is
sensitive,
specific and
available, it is
not an easy task
for a
manufacturer to
go out and go to
China and get
300 hepatitis B
carriers and
undertake the
kind of study
that is being
proposed by
PATH.
So the idea was
to use the
hepatitis B
testing as sort
of a gold
standard,
correlate that
with the
physical
chemical test
like serum
albumin. And in
the future it
would be
possible for
manufacturers
just to use the
simpler test.
Now, that did
not work out
because the
serum albumin
test was not
acceptable
because of
contamination
from the
environment,
because we live
in a sea of the
stuff. But the
principle that
you have a
biological assay
available that
measures exactly
what you're
worried about,
the highest
titer pathogen
hepatitis B and
the major
pathogen, you're
actually lucky
to have that
available to
you. But if that
could be
correlated with
an assay, a much
simpler assay
like the
fluorescein in
the future, then
it might be
possible at a
certain point
when all the
tests have been
validated, to
move over to a
much simpler and
cheaper test.
But conceptually
I would think
the gold
standard would
actually be the
hepatitis B
model, because
you're measuring
exactly what
you're worried
about.
And the second
point I'd like
to make is that
I totally agree
with the Panel's
recommendation
that they should
basically accept
zero detectable
level of
contamination.
My concern is
that that
doesn't go quite
far enough to
answer all the
questions that
need to be
answered.
Because there
exist out there
a number of
detection
systems with
different
outputs and with
different levels
of sensitivity
and specificity.
So, for example,
the fluorescein
test which the
panel seemed to
be interested in
gives you a
readout in terms
of volume. Some
of the DNA tests
for the
hepatitis B
virus give you
an output in
terms of genome
copies. All of
these can
potentially be
useful. So some
kind of
guidance, I
would imagine
Dr. Lin, you
know which one
of those might
be -- some
direction, some
guidance to the
FDA might be
very helpful to
them.
For example,
there's been a
history of
different levels
of sensitivity
and specificity.
In the '60s it
was visible
blood. Then it
was a blood dip
stick. Then it
was an ELISA
test of the
sensitivity and
specificity of
blood ELISAs.
Then it was PCR,
and now it's
fluorescein. And
certainly it's a
moving target.
But basically if
someone said
well there was
no visible blood
on the head,
would that be
acceptable? No.
Would a dip
stick be
acceptable?
Probably no.
Would the
fluorescein and
PCRB and the
best we have for
those two lines
be acceptable?
It's the best we
can do right
now.
So, you know,
some sort of
guidance along
those lines I
would guess
might be helpful
as well to the
FDA.
Thank you very
much.
CHAIRMAN
EDMISTON: Does
the panel have
any questions
for the speaker?
Yes, Dr. Word?
DR. WORD: I
don't have a
question. But
when he started
talking about
the hepatitis B
and how many
copies, I think
if you're
looking for FDA
for the
guidance, you're
looking at
license test
that your agency
itself has
licensed. And if
you get down to
the lower limit
of detectable,
whether it's 200
copies or 150
copies, if it's
200 copies then
it comes in at
199, then but
it's
nondetectable.
But I guess what
I'm saying is
when he was
suggesting that
you have to use
a test because
there's so many,
you know which
ones you've
licensed. So I
don't know, do
we say it has to
be one that's
licensed in the
United States
because that's
the only one
you're going to
look at?
DR. LIN: You're
asking me to
comment? Well,
the one that
licensed, from
our sister
center, Center
for Biologics
and CBER, these
centers their
licensing for
blood donation,
blood donor. But
here the one
that we're
talking about to
assay the
procedure
biocopy; that
you don't have
to use that kind
of blood
licensing
because that's
totally
different
method.
But here I think
that probably
one of the
challenges is
how you assay
those virus
remain on those
top or any fluid
pathway. That is
probably most of
the challenge
that the
manufacturer is
facing. How you
excerpt those
virus out to
assay for those
copying.
CHAIRMAN
EDMISTON: Dr.
David?
MR. DAVID: Thank
you, Mr.
Chairman.
Just to clarify
in my mind what
the Panel voted
on is a question
to the FDA is
the capability
that you have if
you have -- you
kept asking
would should FDA
do if they have
an application
submitted
tomorrow. And in
this kind of
time frame is
the FDA capable
of reproducing
tests to a level
that the Panel
has recommended?
DR. LIN: I
shouldn't have
used the term
"tomorrow." I
don't mean right
away. I mean
just in the near
future we get a
submission. But
the Panel's
recommendation
that will help
us to prepare a
guidance
document to the
industry. That's
essentially what
I mean.
MR. DAVID: Okay.
CHAIRMAN
EDMISTON: Do we
have any further
speakers from
the public?
Dr. Zehrung,
could I call you
to the podium
for a moment?
DR. ZEHRUNG:
Yes.
CHAIRMAN
EDMISTON: This
is more from a
reviewer
perspective, all
right. This
device that you
have that fits
on the end of
your injector?
DR. ZEHRUNG:
Yes.
CHAIRMAN
EDMISTON: The
splash back that
may occur is
contained within
the bottom
chamber, is that
correct? Is this
positioned the
right way on the
device with the
--
DR. ZEHRUNG:
Yes, sir. The
flat portion is
the skin side.
CHAIRMAN
EDMISTON: The
flat portion is
the skin side.
DR. ZEHRUNG: So
that outer
flange would
then contact the
skin.
CHAIRMAN
EDMISTON: Yes.
DR. ZEHRUNG: And
then the tail
end of it,
actually, comes
into close
proximity to the
nozzle of the --
CHAIRMAN
EDMISTON: Okay.
This part right
here. Okay.
DR. ZEHRUNG:
Yes.
CHAIRMAN
EDMISTON: Like
that?
DR. ZEHRUNG:
Right.
CHAIRMAN
EDMISTON: Like a
flying saucer,
correct? Okay.
And so any
potential splash
back is
contained within
this bottom
chamber, is that
correct?
DR. ZEHRUNG:
Both chambers.
CHAIRMAN
EDMISTON: Both
chambers?
DR. ZEHRUNG:
Yes.
CHAIRMAN
EDMISTON: So let
me ask you a
question. It's
contained in
both chambers.
Is there a
membrane that is
above this?
DR. ZEHRUNG:
There is
membrane.
CHAIRMAN
EDMISTON:
There's a
membrane?
DR. ZEHRUNG:
That's --right.
So on the side
that's in close
proximity to the
nozzle --
CHAIRMAN
EDMISTON: Yes.
DR. ZEHRUNG: --
there is a thin
polyethylene
film that the
injection stream
pierces on its
way through the
protector cap
and then into
the tissue.
CHAIRMAN
EDMISTON: Is
that also a
replaceable
membrane or is
that a permanent
membrane?
DR. ZEHRUNG:
It's permanent.
It's actually
welded onto the
backside of the
protector cap
during the
fabrication
process.
CHAIRMAN
EDMISTON: Okay.
And that
membrane
prevents the
splash back from
getting back
into the nozzle
component of the
device?
DR. ZEHRUNG: So
far studies have
indicated that
that that's the
case.
CHAIRMAN
EDMISTON: Okay.
And basically
what you've seen
to date is there
is no -- when
you remove this
membrane and you
look at the --
there obviously
is some residual
material on the
membrane in the
top, correct?
DR. ZEHRUNG:
Sometimes.
CHAIRMAN
EDMISTON:
Sometimes.
DR. ZEHRUNG:
Especially with
the fluorescein
test which is an
exaggerated sort
of test.
CHAIRMAN
EDMISTON: Okay.
But it greatly
reduces to a
significant
extent --
DR. ZEHRUNG:
Definitely.
CHAIRMAN
EDMISTON: -- the
infiltration of
that splash back
into the
nozzles?
DR. ZEHRUNG:
That's true.
CHAIRMAN
EDMISTON: Does
that answer
questions out
there? Okay.
Very good. Thank
you so much.
Do we have any
further comments
or questions?
At this time, I
believe I have
to turn it over
to my Executive
Secretary to
read a
statement.
EXECUTIVE
SECRETARY
COLBURN: Well,
before we
adjourn for the
day, I just want
to remind the
Panel members
that all the
material that
you have
received for
preparation and
review for this
Panel is not
considered
proprietary, and
you do not need
to destroy this
as if it was a
PMA panel or
something like
that. So you are
free to keep the
material that
you have
received.
And I just want
to extend my
thanks and
gratitude for
all the Panel
members, and
invited
consultants from
other panels who
have helped us
out in conveying
for today. And
we appreciate
all your very
informative
comments.
And I would also
like to extend
my thanks to Dr.
Friede from WHO
coming and
presenting to
the Panel, as
well as the
industry
representatives
that have helped
us out in
guiding the
discussions
today and
helping us come
to at least a
consensus and
direction where
we can forward
in developing of
these devices.
Thank you.
I'll return this
back to Dr.
Edmiston for any
final comments.
CHAIRMAN
EDMISTON: And I
also want to
express my
sincere thanks
to the members
of this Panel
for this
diligence and
interest and
effort in
today's
activities. And
the members of
the public,
especially Mr.
Hooks for his
presentation.
And also the
members of
industry for
really a very
interesting
presenting of
what could be
some new
emerging
technology.
If there is no
further
business, I
would like to
adjourn this
meeting of the
General Hospital
and Personal Use
Device Panel.
Thank you very
much.
(Whereupon, the
meeting was
adjourned at
2:41 p.m.)
|