Home >>
Books >>
Hepatology
2012 >>
HTML
#9002:
Hepatology
2012
 14
.
Hepatitis
C:
New
Drugs 14
.
Hepatitis
C:
New
Drugs
Christian
Lange,
Christoph
Sarrazin
Download
the
entire
book
(PDF,
546
pages).
|
|
|
Introduction
Combination
therapy with
pegylated
interferon-a
plus
weight-based
ribavirin
leads to
sustained
virologic
response
(SVR,
defined by
undetectable
HCV RNA 24
weeks after
treatment
completion)
in
approximately
50% of all
HCV genotype
1-infected
patients,
compared to
70-90% of
patients
infected
with HCV
genotypes 2
and 3 (Zeuzem
2009).
The limited
treatment
success of
this
treatment
especially
in HCV
genotype 1
patients,
the long
treatment
durations
(up to 72
weeks), as
well as the
numerous
side-effects
of PEG-IFN α
and
ribavirin
therapy, and
an exploding
knowledge of
the HCV life
cycle and of
structural
features of
the HCV
proteins,
has spurred
the
development
of many
promising
directly
acting
antiviral
agents (DAA)
(Kim 1996,
Lindenbach
2005,
Lohmann
1999, Lorenz
2006, Wakita
2005). In
principle,
each of the
four HCV
structural
and six
non-structural
proteins,
HCV-specific
RNA
structures
such as the IRES, as
well as host
factors on
which HCV
depends, are
suitable
targets for
DAA agents.
In the
following
section, DAA
compounds
currently in
clinical
development
are
presented
(Table 1,
Figure 1).
Table 1.
Selected
directly
acting
antiviral
agents in
the
pipeline.
|
Drug name |
Company |
Target / Active site |
|
Phase |
|
NS3-4A
protease
inhibitors |
|
|
|
|
|
Ciluprevir
(BILN
2061) |
Boehringer
Ingelheim |
Active
site
/
macrocyclic |
|
Stopped |
|
Telaprevir
(VX-950) |
Vertex
/
Janssen |
Active
site
/
linear |
|
IV |
|
Boceprevir
(SCH503034) |
Merck
(S-P) |
Active
site
/
linear |
|
IV |
|
Simeprevir
(TMC435350) |
Janssen
/
Medivir |
Active
site
/ macrocyclic |
also
in
separate
Phase
II
studies
with
other
DAA
as
part
of
IFN-free
regimens,
with
and
without
RBV |
III
No
clinically
significant
interaction
between
the
investigational
HCV
protease
inhibitor
TMC435
and
the
immunosuppressives
cyclosporine
and
tacrolimus
|
|
Danoprevir
(R7227) |
Roche
/
InterMune |
Active
site
/
macrocyclic |
|
II |
|
Vaniprevir
(MK-7009) |
Merck |
Active
site
/
macrocyclic |
|
Halted/II |
|
MK-5172 |
Merck |
Active
site
/
macrocyclic |
|
II |
|
BI201335 |
Boehringer
Ingelheim |
Active
site
/
linear |
|
III |
|
Narlaprevir
(SCH900518) |
Schering-Plough |
Active
site
/
linear |
|
Halted |
|
Asunaprevir
(BMS-650032) |
Bristol-Myers
Squibb |
Active
site |
|
II |
|
PHX1766 |
Phenomix |
Active
site |
|
I |
|
GS-9256 |
Gilead |
Active
site |
|
II |
|
GS-9451 |
Gilead |
Active
site |
|
I |
|
ABT450 |
Abbott |
Active
site |
|
II |
|
IDX320 |
Idenix |
Active
site |
|
II |
|
ACH-1625 |
Achillion |
Active
site
/
macrocyclic? |
|
II |
|
Nucleoside
analog
NS5B
polymerase
inhibitors
Nucleoside
analog
inhibitors
achieves
NS5B
inhibition
NNIs
bind
distantly
to
the
active
centre
of
NS5B,
their
application
may
rapidly
lead
to
the
development
of
resistant
mutants
in
vitro
and
in
vivo.
Moreover,
mutations
at
the
NNI
binding
sites
do
not
necessarily
lead
to
impaired
function
of
the
enzyme.....
active
centre
of
NS5B
is a
highly
conserved
region
of
the
HCV
genome,
NIs
are
potentially
effective
against
different
genotypes.
Single
amino
acid
substitutions
in
every
position
of
the
active
centre
may
result
in
loss
of
function
or
in
extremely
impaired
replicative
fitness.
Thus,
there
is a
relatively
high
barrier
in
the
development
of
resistances
to
NIs.
|
|
Valopicitabine
(NM283) |
Idenix
/
Novartis |
Active
site
|
|
Stopped
gastrointestinal
side-effects |
|
Mericitabine
(R7128)
RO5024048
the
most
advanced
nucleoside
polymerase
inhibitor
all
HCV
genotypes,
and
thus
far
no
viral
resistance
against
mericitabine
|
Roche
/
Pharmasset
|
Active
site
December
2011-March
2014 |
1.
Placebo;
Pegasys;
Copegus;
telaprevir
----------
2.
boceprevir;
Pegasys;
Copegus |
II
Enrollment120
treatment up to 48 weeks |
|
R1626
a
treatment
for
HCV,
molecule
(4'-azidocytidine/PSI-6130
Licensed
PSI-6130
pro-drugs
also
including
R7128
|
Roche
internal
program
to
develop
R1626
for
HCV
treatment |
Active
site
|
R1626
Trial
title
:
Dengue
Completed
A
Study
of
Balapiravir
in
Patients
With
Dengue
Virus
Infection |
Stopped
severe
lymphopenia
and
infectious
disease
adverse
events
Drug:
balapiravir
[RO4588161];
Drug:
placebo |
|
PSI-7977
GS-7977
sofosbuvir NCT01054729
NS5B
Nucleoside
analog
inhibitors-
impaired replicative
fitness NNIs
Qualify
is
based
upon
stratification
for
IL-28b
status; |
Pharmasset/
GSK
NNIs
bind
distantly
to
the
active
centre
of
NS5B |
Active
site
January 21, 2010-May 24, 2012
Last verified: May 2012
|
100
mg
200
mg
400
mg
Placebo
Interferon
(PEG)
Ribavirin (RBV) Eligible patients randomized to one of the 3 active cohorts |
II
twenty (20) subjects for each, with 16 subjects assigned to active PSI-7977 and 4 subjects assigned to matching placebo in a 4:1 randomization.
|
|
PSI-938
demonstrate
high
antiviral
activities
..high
genetic
barrier
to
resistance |
Pharmasset |
Active
site |
|
Stopped
citing
"laboratory
abnormalities
associated
with
liver
function. |
|
IDX184 |
Idenix |
Active
site |
|
|
|
IDX189
BMS-986094 |
|
|
|
Stopped |
|
(HCVet Note:
NEW BL-8020
(replicate)
acts via a
unique
mechanism of
action, by
inhibiting
Hepatitis C
virus
(HCV)-induced autophagy,a
catabolic
process
involving
the
degradation
of a cell's
own
components
through the
lysosomal
machinery,
which
differs from
the
mechanism of
currently
used anti-
HCV agents.
Phase
I/II
clinical
trial
in
Europe
to
test
the
drug's
safety
and
efficacy
during
the
first
quarter
of
2013
BL-8020
inhibits
hepatitis-induced
autophagy
(a
mechanism
by
which
cells
degrade
damaged
or
unnecessary
cellular
components,
including
invading
viruses)
in
the
host
cells,
thereby
reducing
the
ability
of
the
hepatitis
virus
to
replicate...
BiolineRX
has
an
exclusive
worldwide
license
for
BL-8020
from
France
Genoscience
SA.
Non-nucleoside
NS5B
polymerase
inhibitors
(NNI)
non-nucleoside
inhibitors
(NNIs)
...
achieves
NS5B
inhibition
by
binding
to
different
allosteric
enzyme
sites,
which
results
in
conformational
protein
change
before
the
elongation
complex
is
formed
(Beaulieu
2007).
For
allosteric
NS5B
inhibition
high
chemical
affinity
is
required.
NS5B
is
structurally
organized
in a
characteristic
"right
hand
motif",
containing
finger,
palm
and
thumb
domains,
and
offers
at
least
four
NNI-binding
sites,
a
benzimidazole-(thumb
1)-,
thiophene-(thumb
2)-,
benzothiadiazine-(palm
1)-
and
benzofuran-(palm
2)-binding
site
(Lesburg
1999)
(Figure
6).
Because
of
their
distinct
binding
sites,
different
polymerase
inhibitors
can
theoretically
be
used
in
combination
or
in
sequence
to
manage
the
development
of
resistance.
Because
NNIs
bind
distantly
to
the
active
centre
of
NS5B,
their
application
may
rapidly
lead
to
the
development
of
resistant
mutants
in
vitro
and
in
vivo.
Moreover,
mutations
at
the
NNI
binding
sites
do
not
necessarily
lead
to
impaired
function
of
the
enzyme.
Figure
7
shows
the
structure
of
selected
nucleoside
and
non-nucleoside
inhibitors.
|
|
BILB
1941 |
Boehringer
Ingelheim |
NNI
site
1 /
thumb
1 |
|
Stopped |
|
BI207127 |
Boehringer
Ingelheim |
NNI
site
1 /
thumb
1 |
|
II |
|
MK-3281 |
Merck |
NNI
site
1 /
thumb
1 |
|
II |
|
TMC647055 |
Janssen |
NNI
site
1 /
thumb
1 |
|
|
|
Filibuvir
(PF-00868554) |
Pfizer |
NNI
site
2 /
thumb
2 |
|
II |
|
VCH759 |
ViroChem
Pharma |
NNI
site
2 /
thumb
2 |
|
II |
|
VCH916 |
ViroChem
Pharma |
NNI
site
2 /
thumb
2 |
|
II |
|
VCH222 |
ViroChem
Pharma |
NNI
site
2 /
thumb
2 |
|
II |
|
ANA598 |
Anadys |
NNI
site
3 /
palm
1 |
|
II |
|
ABT-072 |
Abbott |
NNI
site
3 /
palm
1 |
|
II |
|
ABT-333 |
Abbott |
NNI
site
3 /
palm
1 |
|
II |
|
HCV-796 |
ViroPharma
/
Wyeth |
NNI
site
4 /
palm
2 |
|
Stopped |
|
GS-9190 |
Gilead |
NNI
site
4 /
palm
2 |
|
II |
|
IDX375 |
Idenix |
NNI
site
4 /
palm
2 |
|
II |
|
NS5A
inhibitor
nucleoside
Daclatasvir
(BMS-790052) |
Bristol-Myers
Squibb |
NS5A
domain
1
inhibitor |
|
II |
|
BMS-824393 |
Bristol-Myers
Squibb |
NS5A
protein |
|
I |
|
PPI-461 |
Presidio
Pharmaceuticals |
NS5A
protein |
|
I |
|
GS-5885 |
Gilead |
NS5A
protein |
|
I |
|
Indirect
inhibitors
/
unknown
mechanism
of
action |
|
|
|
|
Alisporivir
(Debio-025) |
Debiopharm |
Cyclophilin
inhibitor |
|
III |
|
NIM811 |
Novartis |
Cyclophilin
inhibitor |
|
I |
|
SCY-635 |
Scynexis |
Cyclophilin
inhibitor |
|
II |
|
Nitazoxanide |
|
PKR
induction
(?) |
|
II |
|
Miravirsen |
Santaris |
miRNA122
antisense
RNA |
|
II |
|
Celgosivir |
Migenix |
Α-glucosidase
inhibitor |
|
II |
HCV is a
positive-sense
single-stranded
RNA virus of
approximately
9600
nucleotides.
The HCV
genome
contains a
single large
open reading
frame
encoding for
a
polyprotein
of about
3100 amino
acids. From
this
initially
translated
polyprotein,
the
structural
HCV protein
core (C) and
envelope
glycoproteins
1 and 2 (E1,
E2); p7; and
the six
non-structural
HCV proteins
NS2, NS3,
NS4A, NS4B,
NS5A and
NS5B, are
processed by
both viral
and host
proteases.
The core
protein
forms the
viral
nucleocapsid
carrying E1
and E2,
which are
receptors
for viral
attachment
and host
cell entry.
The
non-structural
proteins are
multifunctional
proteins
essential
for the HCV
life cycle (Bartenschlager
2004). P7 is
a small
hydrophobic
protein that
oligomerises
into a
circular
hexamer,
most likely
serving as
an ion
channel
through the
viral lipid
membrane.
The large
translated
section of
the HCV
genome is
flanked by
the strongly
conserved
HCV 3Ž and
5Ž
untranslated
regions (UTR).
The 5ŽUTR is
comprised of
four highly
structured
domains
forming the
internal
ribosome
entry site (IRES),
which plays
an important
role in HCV
replication
(Figure 2).
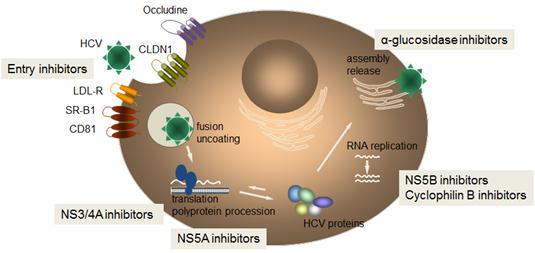
Figure 1.
HCV life
cycle and
targets for
directly
acting
antiviral (DAA)
agents.
After
receptor-mediated
endocytosis,
the fusion
of HCV with
cellular
membranes,
and
uncoating
the viral
nucleocapsid,
the
single-stranded
positive-sense
RNA genome
of the virus
is released
into the
cytoplasm to
serve as a
messenger
RNA for the
HCV
polyprotein
precursor.
HCV mRNA
translation
is under the
control of
the internal
ribosome
entry site (IRES),
formed by
domains
II-IV of the
HCV 5ŽUTR (Moradpour
2007). IRES
mediates HCV
polyprotein
translation
by forming a
stable
complex with
the 40S
ribosomal
subunit,
eukaryotic
initiation
factors and
viral
proteins.
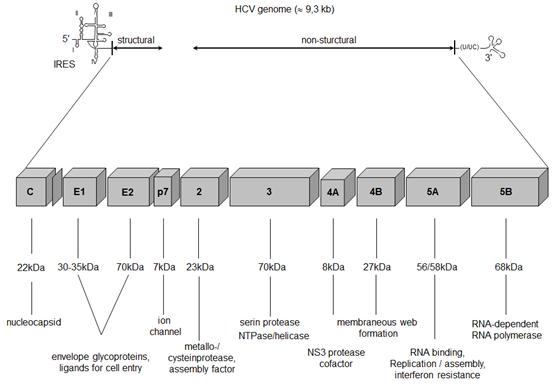
Figure 2.
Genomic
organisation
of HCV.
From the
initially
translated
HCV
polyprotein
the three
structural
and seven
non-structural
HCV proteins
are
processed by
both host
and viral
proteases (Moradpour
2007). NS2
is a
metalloproteinase
that cleaves
itself from
the NS2/NS3
protein,
leading to
its own loss
of function
and to the
release of
the NS3
protein
(Lorenz
2006). NS3
provides a
serine
protease
activity and
a helicase/NTPase
activity.
The serine
protease
domain
comprises
two
b-barrels
and four
α-helices.
The serine
protease
catalytic
triad -
histidine
57,
asparagine
81 and
serine 139 -
is located
in a small
groove
between the
two
b-barrels
(Kim 1996,
Kim 1998).
NS3 forms a
tight,
non-covalent
complex with
its
obligatory
cofactor and
enhancer
NS4A, which
is essential
for proper
protein
folding
(Figure 3).
The NS3-4A
protease
cleaves the
junctions
between
NS3/NS4A,
NS4A/NS4B,
NS4B/NS5A
and
NS5A/NS5B.
Besides its
essential
role in
protein
processing,
NS3 is
integrated
into the HCV
RNA
replication
complex,
supporting
the
unwinding of
viral RNA by
its helicase
activity.
Moreover,
NS3 might
play an
important
role in HCV
persistence
via blocking
TRIF-mediated
toll-like
receptor
signalling
and Cardif-mediated
RIG-I
signalling,
subsequently
resulting in
impaired
induction of
type I
interferons
(Meylan
2005). Thus,
pharmacologic
NS3
inhibition
might
support
viral
clearance by
restoring
the innate
immune
response.
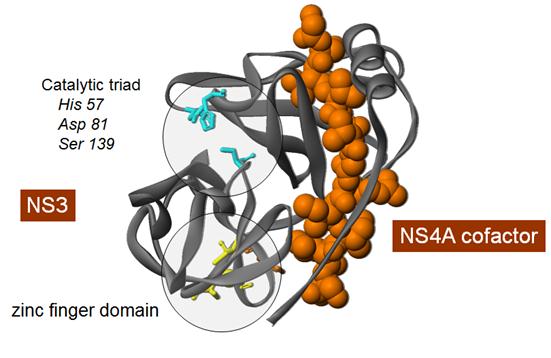
Figure 3.
Molecular
structure of
the HCV
NS3-4A
protease.
The active
site of the
NS3-4A
protease is
located in a
shallow
groove
between the
two
b-barrels of
the protease
making the
design of
compound
inhibitors
relatively
difficult.
Nevertheless,
many NS3-4A
protease
inhibitors
have been
developed
which can be
divided into
two classes,
the
macrocyclic
inhibitors
and linear
tetra-peptide
a-ketoamide
derivatives.
In general,
NS3-4A
protease
inhibitors
have been
shown to
strongly
inhibit HCV
replication
during
monotherapy,
but also may
cause the
selection of
resistant
mutants,
which is
followed by
viral
breakthrough.
The
additional
administration
of pegylated
interferon
and
ribavirin,
however, was
shown to
reduce the
frequency of
development
of
resistance.
Future
strategies
aim for
combination
therapies
with
different
antiviral
drugs to
prevent the
development
of
resistance.
The most
advanced
compounds
are
telaprevir
and
boceprevir,
both
approved in
2011.
The first
clinically
tested
NS3-4A
inhibitor
was
ciluprevir (BILN
2061), an
orally
bioavailable,
peptidomimetic,
macrocyclic
drug binding
non-covalently
to the
active
center of
the enzyme (Lamarre
2003)
(Figure 4).
Ciluprevir
monotherapy
was
evaluated in
a
double-blind,
placebo-controlled
pilot study
in
treatment-naïve
genotype 1
patients
with liver
fibrosis and
compensated
liver
cirrhosis (Hinrichsen
2004).
Ciluprevir,
administered
twice daily
for two days
at doses
ranging from
25 to 500
mg, led to a
mean 2-3 log10
decrease of
HCV RNA
serum levels
in most
patients.
Importantly,
the stage of
disease did
not affect
the
antiviral
efficacy of
ciluprevir.
The
tolerability
and efficacy
of
ciluprevir
in HCV
genotype 2-
and
3-infected
individuals
was examined
in an
equivalent
study
design, but
compared to
genotype 1
patients,
the
antiviral
activity was
less
pronounced
and more
variable in
these
patients (Reiser
2005).
Ciluprevir
development
has been
halted.
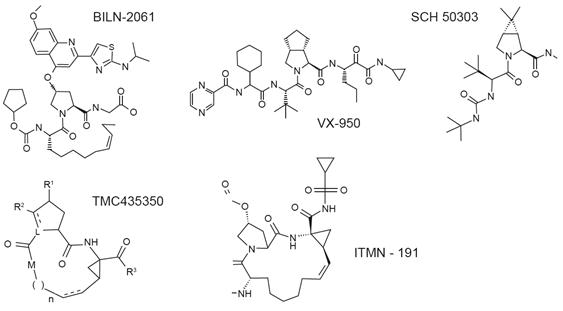
Figure 4.
Molecular
structure of
selected
NS3-4A
inhibitors.
Telaprevir
(Incivek/Incivo®)
and
boceprevir (Victrelis®)
Telaprevir
and
boceprevir
were
approved for
the
treatment of
chronic
hepatitis C
virus
genotype 1
infection by
the FDA, EMA
and several
other
countries in
2011. Both
telaprevir
and
boceprevir
are orally
bioavailable,
peptidomimetic
NS3-4A
protease
inhibitors
belonging to
the class of
a-ketoamid
derivatives
(Figure 4).
Like other
NS3-4A
inhibitors,
telaprevir
and
boceprevir
are
characterized
by a
remarkable
antiviral
activity
against HCV
genotype 1.
However,
monotherapy
with these
agents
results in
the rapid
selection of
drug-resistant
variants
followed by
viral
breakthrough
(Reesink
2006,
Sarrazin
2007). Phase
II and III
clinical
studies have
shown that
the addition
of pegylated
interferon α
plus
ribavirin
leads to a
substantially
reduced
frequency of
resistant
mutants and
viral
breakthrough,
and to
significantly
higher SVR
rates in
both
treatment-naïve
and
treatment-experienced
HCV genotype
1 patients
compared to
treatment
with
pegylated
interferon α
and
ribavirin
alone (Bacon
2011,
Jacobson
2011,
Poordad
2011,
Sherman
2011,
Zeuzem 2011).
Therefore,
telaprevir-
and
boceprevir-based
triple
therapy can
be
considered
the novel
standard of
care for HCV
genotype 1
patients.
Results of
the Phase
III
telaprevir
and
boceprevir
approval
studies are
summarized
in Figure 5.
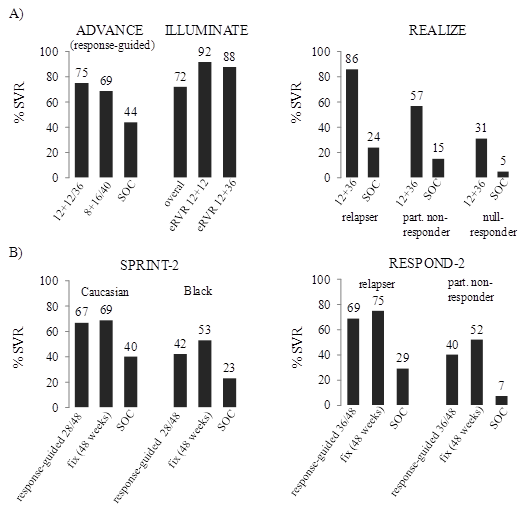
Figure 5.
SVR rates in
Phase III
clinical
trials
evaluating
telaprevir
(A) or
boceprevir
(B) in
combination
with PEG-IFN
α
and
ribavirin.
ADVANCE,
ILLUMINATE
and SPRINT-2
enrolled
treatment-naïve
patients,
REALIZE and
RESPOND-2
enrolled
treatment-experienced
patients.
Telaprevir
was
administered
for 8 or 12
weeks in
combination
with PEG-IFN
α-2a
and
ribavirin,
followed by
12-40 weeks
of PEG-IFN
α-2a
and
ribavirin
alone.
Boceprevir
was
administered
over the
whole
treatment
period of 28
or 48 weeks
in
combination
with PEG-INF
α-2b
and
ribavirin,
except of
the first 4
weeks of
lead-in
therapy of
PEG-IFN
α-2b
and
ribavirin
only. eRVR,
extended
early
virologic
response;
SOC,
standard of
care; LI,
lead-in (4
weeks of
PEG-INF
α
plus
ribavirin
only).
Other NS3
protease
inhibitors
are
currently in
various
phases of
development
(danoprevir
(R7227/ITMN191),
vaniprevir
(MK-7009),
BI201335,
simeprevir
(TMC435350),
narlaprevir
(SCH900518),
asunaprevir
(BMS-650032),
PHX1766,
ACH-1625,
IDX320,
ABT-450,
MK-5172,
GS-9256,
GS-9451) and
will
significantly
increase
treatment
options for
chronic
hepatitis C
in the near
future. In
general,
comparable
antiviral
activities
to
telaprevir
and
boceprevir
in HCV
genotype 1
infected
patients
were
observed
during mono-
(and
triple-)
therapy
studies (Brainard
2010, Manns
2011,
Reesink
2010).
Potential
advantages
of these
second and
third
generation
protease
inhibitors
might be
improved
tolerability,
broader
genotypic
activity,
different
resistance
profiles,
and/or
improved
pharmacokinetics
for
once-daily
dosage
(e.g.,
TMC435,
BI201335).
Different
resistance
profiles
between
linear
tetrapeptide
and
macrocyclic
inhibitors
binding to
the active
site of the
NS3 protease
have been
revealed.
However,
R155 is the
main
overlapping
position for
resistance
and
different
mutations at
this amino
acid site
within the
NS3 protease
confer
resistance
to nearly
all protease
inhibitors
currently in
advanced
clinical
development
(Sarrazin
2010). An
exception is
MK-5172,
which
exhibits
potent
antiviral
activity
against
variants
carrying
mutations at
position
R155. In
addition,
MK-5172 had
potent
antiviral
activity
against both
HCV genotype
1 and 3
isolates (Brainard
2010).
Because of
the high
replication
rate of HCV
and the poor
fidelity of
its
RNA-dependent
RNA
polymerase,
numerous
variants (quasispecies)
are
continuously
produced
during HCV
replication.
Among them,
variants
carrying
mutations
altering the
conformation
of the
binding
sites of DAA
compounds
can develop.
During
treatment
with
specific
antivirals,
these
preexisting
drug-resistant
variants
have a
fitness
advantage
and can be
selected to
become the
dominant
viral
quasispecies.
Many of
these
resistant
mutants
exhibit an
attenuated
replication
with the
consequence
that, after
termination
of exposure
to specific
antivirals,
the wild
type may
displace the
resistant
variants (Sarrazin
2007).
Nevertheless,
HCV
quasispecies
resistant to
NS3-4A
protease
inhibitors
or
non-nucleoside
polymerase
inhibitors
can be
detected at
low levels
in some
patients
(approx. 1%)
who have
never been
treated with
these
specific
antivirals
before (Gaudieri
2009). The
clinical
relevance of
these
pre-existing
mutants is
not
completely
understood,
although
there is
evidence
that they
may reduce
the chance
of achieving
an SVR with
DAA-based
triple
therapies if
the
patient's
individual
sensitivity
to pegylated
interferon α
+ ribavirin
is low.
More
recently,
the Q80R/K
variant has
been
described as
conferring
low-level
resistance
to
simeprevir
(TMC435), a
macrocyclic
protease-inhibitor.
Interestingly,
the Q80K
variant can
be detected
in
approximately
10% of HCV
genotype
1-infected
patients
(typically
in subtype
1a isolates)
and a slower
viral
decline
during
simeprevir-based
triple
therapy was
observed
(Lenz 2011).
Table 2
summarizes
the
resistance
profile of
selected
NS3-4A
inhibitors.
Although the
resistance
profiles
differ
significantly,
R155 is an
overlapping
position for
resistance
development
and
different
mutations at
this
position
confer
resistance
to nearly
all protease
inhibitors
(not
MK-5172)
currently in
advanced
clinical
development
(Sarrazin
2010).
Importantly,
many
resistance
mutations
could be
detected
in vivo
only by
clonal
sequencing.
For example,
mutations at
four
positions
conferring
telaprevir
resistance
have been
characterized
so far
(V36A/M/L,
T54A,
R155K/M/S/T
and
A156S/T),
but only
A156 could
be
identified
initially
in vitro
in the
replicon
system (Lin
2005). These
mutations,
alone or as
double
mutations,
conferred
low (V36A/M,
T54A,
R155K/T,
A156S) to
high
(A156T/V,
V36M +
R155K, V36M
+ 156T)
levels of
resistance
to
telaprevir (Sarrazin
2007). It is
thought that
the
resulting
amino acid
changes of
these
mutations
alter the
confirmation
of the
catalytic
pocket of
the
protease,
which
impedes
binding of
the protease
inhibitor (Welsch
2008).
Table 2.
Resistance
mutations to
HCV NS3
protease
inhibitors.
|
|
36 |
54 |
55 |
80 |
155 |
156A |
156B |
168 |
170 |
|
Telaprevir*
(linear) |
|
|
|
|
|
|
|
|
|
|
Boceprevir*
(linear) |
|
|
|
|
|
|
|
|
|
|
SCH900518*
(linear) |
|
|
|
|
|
|
|
|
|
|
BI-201335*
(linear?) |
|
|
|
|
|
|
|
|
|
|
BILN-2061
**
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
Danoprevir*
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
MK-7009*
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
TMC435*
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
BMS-650032* |
|
|
|
|
|
|
|
|
|
|
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
GS-9451* |
|
|
|
|
|
|
|
|
|
|
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
ABT450* |
|
|
|
|
|
|
|
|
|
|
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
IDX320** |
|
|
|
|
|
|
|
|
|
|
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
ACH1625** |
|
|
|
|
|
|
|
|
|
|
(macrocyclic) |
|
|
|
|
|
|
|
|
|
|
MK-5172*** |
|
|
|
|
|
|
|
|
|
|
(macrocyclic) |
|
|
|
|
|
|
|
|
|
* mutations
associated
with
resistance
in patients
** mutations
associated
with
resistance
in vitro
*** no viral
break-through
during 7
days
monotherapy
# Q80
variants
have been
observed in
approximately
10% of
treatment-naïve
patients and
was
associated
with slower
viral
decline
during
simeprevir
(TMC435)
triple
therapy
As shown for
other NS3-4A
protease
inhibitors
as well
(e.g.,
danoprevir),
the genetic
barrier to
telaprevir
resistance
differs
significantly
between HCV
subtypes. In
all clinical
studies of
telaprevir
alone or in
combination
with PEG-IFN
α and
ribavirin,
viral
resistance
and
breakthrough
occurred
much more
frequently
in patients
infected
with HCV
genotype 1a
compared to
genotype 1b.
This
difference
was shown to
result from
nucleotide
differences
at position
155 in HCV
subtype 1a (aga,
encodes R)
versus 1b (cga,
also encodes
R). The
mutation
most
frequently
associated
with
resistance
to
telaprevir
is R155K;
changing R
to K at
position 155
requires 1
nucleotide
change in
HCV subtype
1a, and 2
nucleotide
changes in
subtype 1b
isolates (McCown
2009).
It will be
important to
define
whether
treatment
failure due
to the
development
of variants
resistant to
DAA agents
has a
negative
impact on
re-treatment
with the
same or a
different
DAA
treatment
regimen.
Follow-up
studies of
telaprevir
and
boceprevir
Phase III
studies have
revealed a
rapid
decline of
resistant
variants
below the
limit of
detection
(>20% of
quasispecies)
of
population
sequencing
techniques
(Barnard
2011,
Sherman
2011).
However,
telaprevir-
and
boceprevir-resistant
variants
were
detectable
by a clonal
sequencing
approach
several
years after
treatment in
single
patients who
had been
treated with
telaprevir
or
boceprevir
within
smaller
Phase Ib
studies (Susser
2011).
HCV
replication
is initiated
by the
formation of
the
replication
complex, a
highly
structured
association
of viral
proteins and
RNA, of
cellular
proteins and
cofactors,
and of
rearranged
intracellular
lipid
membranes
derived from
the
endoplasmic
reticulum (Moradpour
2007). The
key enzyme
in HCV RNA
replication
is NS5B, an
RNA-dependent
RNA
polymerase
that
catalyzes
the
synthesis of
a
complementary
negative-strand
RNA by using
the
positive-strand
RNA genome
as a
template (Lesburg
1999)
(Figure 6).
From this
newly
synthesized
negative-strand
RNA,
numerous RNA
strands of
positive
polarity are
produced by
NS5B
activity
that serve
as templates
for further
replication
and
polyprotein
translation.
Because of
poor
fidelity
leading to a
high rate of
errors in
its RNA
sequencing,
numerous
different
isolates are
generated
during HCV
replication
in a given
patient,
termed HCV
quasispecies.
It is
reasoned
that due to
the lack of
proofreading
of the NS5B
polymerase
together
with the
high
replication
of HCV,
every
possible
mutation is
generated
each day.
NS5B
RNA
polymerase
inhibitors
can be
divided into
two distinct
categories.
Nucleoside
analog
inhibitors (NIs)
like
valopicitabine
(NM283),
mericitabine
(R7128),
R1626,
PSI-7977,
PSI-938 or
IDX184 mimic
the natural
substrates
of the
polymerase
and are
incorporated
into the
growing RNA
chain, thus
causing
direct chain
termination
by tackling
the active
site of NS5B
(Koch 2006).
Because the
active
centre of
NS5B is a
highly
conserved
region of
the HCV
genome, NIs
are
potentially
effective
against
different
genotypes.
Single amino
acid
substitutions
in every
position of
the active
centre may
result in
loss of
function or
in extremely
impaired
replicative
fitness.
Thus, there
is a
relatively
high barrier
in the
development
of
resistances
to NIs.
In contrast
to NIs, the
heterogeneous
class of
non-nucleoside
inhibitors (NNIs)
achieves
NS5B
inhibition
by binding
to different
allosteric
enzyme
sites, which
results in
conformational
protein
change
before the
elongation
complex is
formed
(Beaulieu
2007). For
allosteric
NS5B
inhibition
high
chemical
affinity is
required.
NS5B is
structurally
organized in
a
characteristic
"right hand
motif",
containing
finger, palm
and thumb
domains, and
offers at
least four
NNI-binding
sites, a
benzimidazole-(thumb
1)-,
thiophene-(thumb
2)-,
benzothiadiazine-(palm
1)- and
benzofuran-(palm
2)-binding
site (Lesburg
1999)
(Figure 6).
Because of
their
distinct
binding
sites,
different
polymerase
inhibitors
can
theoretically
be used in
combination
or in
sequence to
manage the
development
of
resistance.
Because NNIs
bind
distantly to
the active
centre of
NS5B, their
application
may rapidly
lead to the
development
of resistant
mutants
in vitro
and in
vivo.
Moreover,
mutations at
the NNI
binding
sites do not
necessarily
lead to
impaired
function of
the enzyme.
Figure 7
shows the
structure of
selected
nucleoside
and
non-nucleoside
inhibitors.
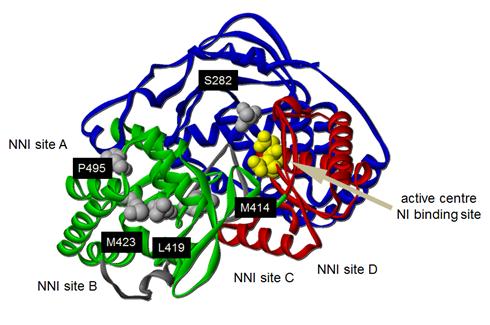
Figure 6.
Structure of
the HCV NS5B
RNA
polymerase
and binding
sites.

Figure 7.
Molecular
structure of
selected
NS5B
polymerase
inhibitors.
Valopicitabine
(NM283,
2'-C-methylcytidine/NM107),
the first
nucleoside
inhibitor
investigated
in patients
with chronic
hepatitis C,
showed a low
antiviral
activity (Afdhal
2007). Due
to
gastrointestinal
side effects
the clinical
development
of NM283 was
stopped.
The second
nucleoside
inhibitor to
be reported
in patients
with chronic
hepatitis C
was R1626
(4'-azidocytidine/PSI-6130).
A Phase 1
study in
genotype
1-infected
patients
observed a
high
antiviral
activity at
high doses
of R1626 in
genotype
1-infected
patients (Pockros
2008). No
viral
breakthrough
with
selection of
resistant
variants was
reported
from
monotherapy
or
combination
studies with
pegylated
interferon ±
ribavirin (Pockros
2008). Due
to severe
lymphopenia
and
infectious
disease
adverse
events
further
development
of R1626 was
stopped.
Mericitabine
(RG7128) is
still in
development
and the most
advanced
nucleoside
polymerase
inhibitor.
Mericitabine
is safe and
well-tolerated,
effective
against all
HCV
genotypes,
and thus far
no viral
resistance
against
mericitabine
has been
observed in
clinical
studies.
Interim
results of
current
Phase II
clinical
trials in
HCV genotype
1-, 2-,
3-infected
patients of
R7128 in
combination
with
pegylated
interferon
and
ribavirin
revealed
superior SVR
rates of
mericitabine-based
triple
therapy
compared to
PEG-IFN α
alone (Pockros
2011). In an
all oral
regimen,
administration
of R7128 in
combination
with the
protease
inhibitor
R7227/ITMN191
for 14 days,
a
synergistic
antiviral
activity of
both drugs
was observed
(Gane 2010).
No viral
breakthrough
with
selection of
resistant
variants has
been
reported.
Very
promising
clinical
data have
been
published
recently for
PSI-7977, a
nucleoside
analog NS5B
inhibitor
effective
against all
HCV
genotypes.
In HCV
genotype 2-
and 3-
infected
patients,
PSI-7977
(400 mg once
daily) in
combination
with
ribavirin
for 12 weeks
+ PEG-IFN
α for
4-12 weeks
resulted in
100% RVR and
100% week 12
SVR rates (Gane
2011). No
PSI-7977-associated
side effects
have been
reported,
and no
virologic
breakthrough
has been
observed. A
second study
evaluated
PSI-7977-based
triple
therapy in
treatment-naïve
HCV genotype
1-infected
patients. In
this study,
PSI-7977 was
administered
for 12
weeks,
together
with PEG-IFN
α and
ribavirin
for 24 or 48
weeks in
total,
according to
whether HCV
RNA was
below the
limit of
detection at
treatment
weeks 4 and
12 or not,
respectively
(Lawitz
2011). Most
patients
were
eligible for
the
shortened
treatment
duration of
24 weeks,
and SVR was
achieved in
approximately
90% of all
patients.
Other
nucleoside
analogs
(e.g.,
PSI-938 and
IDX184) are
at earlier
stages of
clinical
development
(Sarrazin
2010).
Overall, the
newer
nucleoside
analogs
(PSI-7977,
PSI-938)
also
demonstrate
high
antiviral
activities
that,
together
with their
high genetic
barrier to
resistance,
suggest that
they are
optimal
candidates
for all-oral
combination
therapies
(see below).
At
least 4
different
allosteric
binding
sites have
been
identified
for
inhibition
of the NS5B
polymerase
by
non-nucleoside
inhibitors.
Currently,
numerous
non-nucleoside
inhibitors
are in Phase
I and II
clinical
evaluation
(e.g., NNI
site 1
inhibitor
BI207127;
NNI site 2
inhibitors
filibuvir
(PF-00868554),
VCH-759,
VCH-916 and
VCH-222; NNI
site 3
inhibitor
ANA598, NNI
site 4
inhibitors
HCV-796, and
ABT-333)
(Ali 2008,
Cooper 2007,
Erhardt
2009,
Kneteman
2009). In
general,
these
non-nucleoside
analogs
display a
low to
medium
antiviral
activity and
a low
genetic
barrier to
resistance,
evidenced by
frequent
viral
breakthrough
during
monotherapy
studies and
selection of
resistance
mutations at
variable
sites of the
enzyme. In
line with
these
experiences
in Phase I
studies, a
Phase II
triple
therapy
study with
filibuvir in
combination
with
pegylated
interferon
and
ribavirin
showed high
relapse and
relative low
SVR rates
(Jacobson
2010). In
contrast to
nucleoside-analogs,
non-nucleoside
analogs in
general do
not display
antiviral
activity
against
different
HCV
genotypes (Sarrazin
2010). Due
to their low
antiviral
efficacy and
low genetic
barrier to
resistance,
non-nucleoside
analogs will
probably not
be developed
as part of
triple
therapy but
rather as
components
of quadruple
or all-oral
regimens
(see below).
The HCV NS5A
protein
seems to
play a
manifold
role in HCV
replication,
assembly and
release (Moradpour
2007). It
was shown
that NS5A is
involved in
the early
formation of
the
replication
complex by
interacting
with
intracellular
lipid
membranes,
and it
initiates
viral
assembly at
the surface
of lipid
droplets
together
with the HCV
core (Shi
2002). NS5A
may also
serve as a
channel that
helps to
protect and
direct viral
RNA within
the
membranes of
the
replication
complex (Tellinghuisen
2005).
Moreover, it
was
demonstrated
that NS5A is
able to
interact
with NS5B,
which
results in
an enhanced
activity of
the HCV RNA
polymerase.
Besides its
regulatory
impact on
HCV
replication,
NS5A has
been shown
to modulate
host cell
signaling
pathways,
which has
been
associated
with
interferon
resistance (Wohnsland
2007).
Furthermore,
mutations
within the
NS5A protein
have been
clinically
associated
with
resistance /
sensitivity
to IFN-based
antiviral
therapy (Wohnsland
2007).
BMS-790052
was the
first NS5A
inhibitor to
be
clinically
evaluated.
Even low
doses of
BMS-790052
display high
antiviral
efficacy
against all
HCV
genotypes
in vitro.
Monotherapy
with
BMS-790052
led to a
sharp
initial
decline of
HCV RNA
concentrations,
though its
genetic
barrier to
resistance
is
relatively
low (Gao
2010).
According to
an interim
analysis of
a Phase IIb
clinical
trial in
treatment-naïve
HCV genotype
1 and 4
patients,
treatment
with 20 or
60 mg
BMS-790052
once daily
in
combination
with PEG-IFN
α and
ribavirin
for 24 or 28
weeks, 54%
of all
patients
achieved an
extended RVR,
compared to
13% in the
control
group (Hezode
2011). SVR
rates of
this study
are awaited.
During
monotherapy,
rapid
selection of
variants
resistant to
BMS-790052
occurred
(Nettles
2011). The
most common
resistance
mutations in
HCV genotype
1a patients
were
observed at
residues
M28, Q30,
L31, and Y93
of NS5A. In
HCV genotype
1b patients,
resistance
mutations
were
observed
less
frequently,
predominantly
at positions
L31 and Y93.
These
resistance
mutations
increased
the EC50 to
BMS-790052
moderately
to strongly
(Fridell
2011).
However, no
cross-resistance
between
BMS-790052
and other
DAA agents
has been
reported.
Collectively,
BMS-790052
is a highly
promising
agent for
both triple
therapy as
well as all-DAA
combination
therapy
approaches.
Other NS5A
inhibitors
(e.g.,
BMS-824393,
PPI-461,
GS-5885) are
in early
clinical
development.
The
tetraspanin
protein
CD81,
claudin-1,
occludine,
scavenger
receptor
class B type
1 (SR-B1),
the
low-density
lipoprotein
(LDL)
receptor,
glycosaminoglycans
and the
dendritic
cell- /lymph
node-specific
intercellular
adhesion
molecule
3-grabbing
non-integrin
(DC-SIGN/L-SIGN)
have been
identified
as putative
ligands for
E1 and E2 in
the viral
attachment
and entry
steps (Moradpour
2007). HCV
entry
inhibition
might enrich
future
hepatitis C
treatment
opportunities,
in
particular
in the
prevention
of HCV liver
graft
reinfection.
HCV entry
inhibition
can be
theoretically
achieved by
the use of
specific
antibodies
or small
molecule
compounds
either
blocking E1
and E2 or
their
cellular
receptors.
So far, only
results from
clinical
trials using
polyclonal
(e.g.,
civacir)
(Davis 2005)
or
monoclonal
(e.g.,
HCV-AB 68) (Schiano
2006)
HCV-specific
antibodies
are
available.
The clinical
benefit of
these
antibodies
has been
poor,
however. The
development
of small
molecule
entry
inhibitors
is in a
preclinical
stage and is
complicated
by
difficulties
in the
crystallographic
characterization
of HCV
envelope
proteins.
HCV depends
on various
host factors
throughout
its life
cycle.
Cyclophilin
B is
expressed in
many human
tissues and
provides a
cis-trans
isomerase
activity,
which
supports the
folding and
function of
many
proteins.
Cyclophilin
B enhances
HCV
replication
by
incompletely
understood
mechanisms,
like the
modulation
of NS5B
activity.
Debio-025 (alisporivir)
is an orally
bioavailable
cyclophilin
B inhibitor
exerting an
antiviral
impact on
both HCV and
HIV
replication.
In clinical
trials in
HIV- and
HCV-coinfected
patients,
treatment
with 1200 mg
Debio-025
twice daily
for two
weeks led to
a mean
maximal log10
reduction of
HCV RNA of
3.6 and of
HIV DNA of
1.0 (Flisiak
2008).
Debio-025
was
well-tolerated
and no viral
breakthrough
occurred
during the
14 days of
treatment.
Combination
therapy of
Debio-025
200 mg, 600
mg or 1000
mg and PEG-IFN
a-2a
was
evaluated in
a
double-blind
placebo-controlled
Phase II
trial in
treatment-naïve
patients
monoinfected
with HCV
genotypes 1,
2, 3 or 4.
Treatment
was
administered
for 29 days.
Mean log10
reductions
in HCV RNA
at day 29
were 4.75
(1000 mg),
4.61 (600
mg) and 1.8
(200 mg) in
the
combination
therapy
groups
compared to
2.49 (PEG-IFN
a-2a
alone) and
2.2 (1000 mg
Debio-025
alone) in
the
monotherapy
groups. No
differences
in antiviral
activity
were
observed
between
individuals
infected
with the
different
genotypes.
Debio-025
was safe and
well
tolerated
but led to a
reversible
bilirubin
increase in
some of the
patients
treated with
1000 mg
Debio-025
daily (Flisiak
2009). A
high genetic
barrier to
resistance
of Debio-025
and a broad
HCV
genotypic
activity
highlight
the
potential of
drugs
targeting
host
proteins.
In a Phase
II clinical
trial in
treatment-naïve
HCV genotype
1 patients,
combination
therapy with
Debio-025,
PEG-IFN
a-2a
and
ribavirin
for 24-48
weeks
resulted in
SVR rates of
69-76%
compared to
55% in the
control
group (Flisiak
2011).
Nitazoxanide
with its
active
metabolite
tizoxanide
is a
thiazolide
antiprotozoal
approved for
the
treatment of
Giardia
lamblia
and
Cryptosporidium
parvum
infections.
In vitro
studies have
revealed an
essential
inhibitory
impact on
HCV and HBV
replication
by still
unknown
mechanisms.
Results of
two Phase 2
studies
evaluating
500 mg
nitazoxanide
twice daily
for 12 weeks
followed by
nitazoxanide,
PEG-IFN α-2a
± RBV for 36
weeks
yielded
conflicting
results with
SVR rates of
79% in
treatment-naïve
genotype 4
patients,
but of only
44% in HCV
genotype 1
patients (Rossignol
2009).
Additional
studies are
warranted to
determine
the role of
nitazoxanide
in the
treatment of
chronic
hepatitis C.
Silymarin,
an extract
of milk
thistle (Silybum
marianum)
with
antioxidant
activity,
has been
empirically
used to
treat
chronic
hepatitis C
and other
liver
diseases.
Silibinin is
one of the
six major
flavonolignans
in silymarin.
Surprisingly,
recent
reports
demonstrated
that
silibinin
inhibits HCV
at various
steps of its
life cycle
(Ahmed-Belkacem
2010,
Wagoner
2010). In
addition,
intravenous
silibinin in
non-responders
to prior IFN-based
antiviral
therapy led
to a decline
in HCV RNA
between 0.55
to 3.02 log10
IU/ml after
7 days and a
further
decrease
after an
additional 7
days in
combination
with PEG-IFN
α-2a/RBV in
the range of
1.63 and
4.85 log10
IU/ml (Ferenci
2008).
Ongoing
studies will
clarify the
role of
silibinin in
the
treatment of
chronic
hepatitis C,
including
HCV liver
graft
reinfection.
MicroRNA-122
(miRNA-122)
is a
liver-specific
microRNA
that has
been shown
to be a
critical
host factor
for HCV (Landford
2010).
MiRNA-122
binds to the
5ŽNTR region
of the HCV
genome,
which
appears to
be vital in
the HCV
replication
process.
Miravirsen
is a
modified
antisense
oligonucleotide
that targets
miRNA-122
and thereby
prevents
binding of
miRNA-122 to
the HCV
genome. In a
Phase IIa
proof-of-principle
study,
weekly
subcutaneous
injections
of
miravirsen
led to a
reduction of
HCV RNA
serum
concentration
of up to 2.7
log10
IU/mL,
indicating
that an
antisense
oligonucleotide-based
approach of
miRNA-122
inhibition
could be a
promising
modality for
antiviral
therapy
(Janssen
2010). No
relevant
side effects
were seen in
this study.
The approval
of the HCV
protease
inhibitors
telaprevir
and
boceprevir
in 2011
constitutes
a milestone
in the
treatment of
chronic HCV
genotype 1
infection.
Nevertheless,
telaprevir-
or
boceprevir-based
triple
therapy has
certain
limitations.
In
particular,
treatment
success
still
depends on
the
interferon-sensitivity
of
individual
patients
because a
slow decline
of HCV viral
load during
triple
therapy is
associated
with a high
risk of
antiviral
resistance
development.
Consequently,
viral
breakthrough
of drug
resistant
variants was
observed in
a
significant
number of
patients
with partial
or null
response to
previous
treatment
with PEG-IFN
α and
ribavirin,
in patients
with limited
decline of
HCV viral
load during
lead-in
treatment
with PEG-IFN
α and
ribavirin
alone, or in
difficult to
cure
populations
like Blacks
or patients
with
advanced
liver
fibrosis. In
addition,
triple
therapy is
not an
option for
patients
with
contraindications
to PEG-IFN α
or
ribavirin,
such as
patients
with
decompensated
liver
cirrhosis or
liver
transplant
failure.
To overcome
these
limitations,
numerous
trials have
been
initiated to
investigate
the
potential of
combination
therapies
with
different
DAA agents
alone (Table
3). As is
well
established
in the
treatment of
HIV
infection,
combining
DAA agents
with
different
antiviral
resistance
profiles
should
result in a
substantially
decreased
risk of
viral
breakthrough
of resistant
variants.
Nucleoside
analog NS5B
inhibitors
plus drugs
targeting
host factors
such as the
cyclophilin
inhibitor
alisporivir
display a
high genetic
barrier to
resistance
development
and may
therefore be
key agents
for
effective
DAA
combination
therapies (Sarrazin
2010). In
contrast,
NS3-4A and
NS5A
inhibitors
display a
low genetic
barrier to
resistance
development,
but in view
of their
high
antiviral
efficacy
they appear
to be
promising
combination
partners for
nucleoside
analogs or
cyclophilin
inhibitors.
Due to their
low
antiviral
efficacy and
low genetic
barrier to
resistance
development,
the role of
non-nucleoside
analog NS5B
inhibitors
is currently
less clear.
A potential
advantage of
non-nucleoside
analogs is
their
binding to
multiple
target sites
that may
allow
simultaneous
treatment
with several
non-nucleoside
analogs.
Currently,
DAA
combination
treatment
regimens can
be
classified
according to
the usage of
PEG-IFN α
into
quadruple
therapy
regimens and
all-oral
therapy
regimens.
Quadruple
therapy
approaches
are based on
therapy of
PEG-IFN α
and
ribavirin
plus
combination
of two DAA
agents from
different
classes. In
contrast,
all-oral
treatment
comprises
interferon-free
regimens
including
combinations
of various
DAA
compounds
with or
without
ribavirin.
Preliminary
SVR data of
a small but
highly
informative
trial serves
as a
proof-of-concept
for the
potential of
quadruple
therapy
approach for
patients
with
previous
null
response to
PEG-IFN α +
ribavirin (Lok
2011). In
this Phase
II study, 11
HCV genotype
1 patients
with prior
null
response
were treated
with a
combination
of the NS5A
inhibitor
BMS-790052
and the
protease
inhibitor
BMS-650032
together
with PEG-IFN
α and
ribavirin
for 24
weeks.
Quadruple
therapy
resulted in
100% SVR 12
weeks after
treatment
completion
in both HCV
genotype 1a-
and
1b-infected
patients.
Even though
the number
of patients
included in
this trial
was very
limited,
this high
SVR rate
after
quadruple
therapy
seems
impressive
compared to
SVR rates of
~30% that
were
achieved
with
telaprevir-based
triple
therapy in
prior null
responders (Zeuzem
2011).
A Phase II
clinical
trial
assessed
quadruple
therapy with
the
non-nucleoside
NS5B
inhibitor
tegobuvir in
combination
with the
NS3-4A
inhibitor
GS-9256 +
PEG-IFN α
and
ribavirin
for 28 days
in
treatment-naïve
HCV genotype
1 patients (Zeuzem
2011).
The primary
endpoint of
this study
was rapid
virologic
response (RVR),
which was
achieved in
100% of
patients.
After 28
days of
quadruple
therapy,
treatment
with PEG-IFN
α and
ribavirin
was
continued,
which led to
complete
early
virologic
reponse (cEVR)
in 94% of
patients (Zeuzem
2011).
Another
Phase II
clinical
trial
investigated
a
response-guided
approach
during
quadruple
therapy
containing
the
non-nucleoside
NS5B
inhibitor
VX-222 (100
mg or 400
mg) in
combination
with the
NS3-4A
inhibitor
telaprevir +
PEG-IFN α
and
ribavirin in
treatment-naïve
HCV genotype
1 patients
(Nelson
2011).
Quadruple
treatment
was
administered
for 12
weeks. All
treatment
was stopped
after 12
weeks in
patients who
were HCV
RNA-negative
at treatment
weeks 2 and
8. Patients
in whom HCV
RNA was
detectable
at treatment
week 2 or 8
received an
additional
12 weeks of
PEG-IFN α
and
ribavirin
alone. Up to
50% of
patients met
the criteria
for the
12-week
treatment
duration. Of
those,
82-93%
achieved an
SVR 12 weeks
after
treatment
completion.
In patients
who were
treated with
an
additional
12 weeks of
PEG-IFN α
and
ribavirin,
the
end-of-treatment
response was
100%.
Collectively,
the
quadruple
therapy
approach
appears to
be highly
promising in
patients
with limited
sensitivity
to
interferon-α,
even in
patients
with HCV
subtype 1a.
A first
interferon-free
clinical
trial (the
INFORM-1
study)
evaluated
the
combination
of a
polymerase
inhibitor
(R7128) and
an NS3
inhibitor
(R7227/ITMN191).
In this
proof of
principle
study,
patients
were treated
with both
compounds
for up to 2
weeks (Gane
2010). HCV
RNA
concentrations
decreased by
up to 5.2
log10
IU/ml, viral
breakthrough
was observed
in only one
patient
(although no
resistant
HCV variants
were
identified),
and HCV RNA
was
undetectable
at the end
of dosing in
up to 63% of
treatment-naïve
patients.
However, the
fundamental
question of
whether an
SVR can be
achieved
with
combination
therapies of
different
DAA
compounds
without PEG-IFN
α and
ribavirin
was not
answered by
this trial.
SVR data are
available
for a Phase
II clinical
trial
investigating
therapy with
the NS5A
inhibitor
BMS-790052
in
combination
with the
NS3-4A
protease
inhibitor
BMS-60032
for 24 weeks
in 10 HCV
genotype 1
patients
with a
previous
null
response to
PEG-IFN α
and
ribavirin (Lok
2011). 36%
of patients
achieved an
SVR 24 weeks
after
treatment
completion.
All patients
with viral
breakthrough
were
infected
with HCV
genotype 1a,
and in all
of them HCV
variants
with
resistance
mutations
against both
agents were
detected.
Although
data of
longer
follow-up
periods are
needed, this
trial
constitutes
a
proof-of-principle
that SVR can
be achieved
via all-oral
regimens,
even in
patients
infected
with HCV
subtype 1b.
This was
confirmed
with a 100%
SVR rate in
a small
study
evaluating
the same
agents
(BMS-790052
and
BMS-60032)
in Japanese
HCV genotype
1b previous
null
responders (Chayama
2011).
Another
trial has
investigated
12 weeks of
PSI-7977
monotherapy
(400 mg once
daily) in
HCV genotype
2- and
3-infected
patients
(n=10). 100%
of patients
achieved an
RVR and EOTR,
which
translated
into an SVR
in 60% of
patients (Gane
2011).
Two trials
evaluated
all-oral DAA
combination
therapies
with
ribavirin.
In one of
them,
combination
therapy of
the NS3-4A
inhibitor
BI-201335,
the
non-nucleoside
NS5B
inhibitor
BI-207127
(400 or 600
mg TID) and
ribavirin
for 4 weeks
was assessed
(Zeuzem
2011).
Virologic
response
rates in
patients
treated with
600 mg TID
of BI-207127
were 82%,
100% and
100% at
treatment
days 15, 22,
and 29,
respectively
(Zeuzem
2011).
In patients
who received
the lower
dose of
BI-207127,
virologic
response
rates were
significantly
lower, and
in these
patients
lower
virologic
response
rates were
observed for
patients
infected
with HCV
subtype 1a
compared to
subtype 1b.
Another
trial
compared
tegobuvir (a
non-nucleoside
NS5B
inhibitor) +
GS-9256 (a
NS3-4A
inhibitor)
with or
without
ribavirin in
treatment-naïve
HCV genotype
1 patients (Zeuzem
2011).
Importantly,
tegobuvir +
GS-9256 +
ribavirin
led to a
higher HCV
RNA decline
after 28
days of
treatment
compared to
tegobuvir +
GS-9256
alone (-5.1
log10
vs. -4.1 log10,
respectively),
indicating
that
ribavirin
might be an
important
component of
interferon-free
DAA
combination
therapies.
SVR data of
these and
additional
combination
therapy
regimens are
expected in
the near
future.
Additional
trials
investigated
all-oral
combination
regimens
with
ribavirin in
HCV genotype
2 and 3
patients. 12
weeks of
PSI-7977
plus
ribavirin
resulted in
100% RVR,
EOTR, and
SVR rates in
a small
number of
treatment-naïve
patients
(n=10) (Gane
2011). In
contrast,
during
treatment
with the
cyclophilin
A inhibitor
alisporivir
in
combination
with
ribavirin,
only
approximately
50% of HCV
genotype 2
and 3
patients
became HCV
RNA-negative
at treatment
week 6 (Pawlotsky
2011).
Nevertheless,
these data
highlight
the
impressive
potential of
all-oral
regimens,
when agents
with little
risk of
antiviral
resistance
development
such as
nucleoside
analog NS5B
inhibitors
are used in
combination
with
ribavirin.
Table 3.
Selected
trials
evaluating
DAA
combination
therapies.
|
DAAs
combined |
Additional
medication |
Phase |
|
BMS-650032
(NS3-4A
inhibitor) |
+ /
-
PEG-IFN
α
and
ribavirin |
II |
|
+
BMS-790052
(N5A
inhibitor) |
|
BI-201335
(NS3-4A
inhibitor) |
+
ribavirin
+ /
-
PEG-IFN
α
|
II |
|
+
BI-207127
(non-nuc.
NS5B
inhibitor) |
|
GS-9190
(non-nuc.
NS5B
inhibitor) |
+ /
-
ribavirin
+ /
-
PEG-IFN
α |
II |
|
+
GS-92568
(NS3-4A
inhibitor) |
|
Danoprevir
(NS3-4A
inhibitor) |
followed
by
PEG-IFN
α
and
ribavirin |
II |
|
+
RG-7128
(nuc.
NS5B
inhibitor) |
|
Telaprevir
(NS3-4A
inhibitor) |
+ /
-
ribavirin
+ /
-
PEG-IFN
α |
II |
|
+
VX-222
(non-nuc.
NS5B
inhibitor) |
|
PSI-938
(purine
nuc.
NS5B
inhibitor) |
- |
II |
|
+
PSI-7977
(pyrimidine
nuc.
NS5B
inhibitor) |
Over the
last years,
attempts
have been
made to
reduce side
effects and
treatment
discomfort
of PEG-IFN
α. However,
interferons
with
longer
half-life
and
sustained
plasma
concentrations
(e.g.,
albinterferon,
a fusion
protein of
IFN
α 2b
with human
albumin)
have so far
shown no
overall
benefit with
respect to
SVR rates (Zeuzem
2010). Still
promising is
the
development
of pegylated
interferon
lambda 1
(PEG-IFN
lambda 1).
Like other
type 3
interferons,
IFN lambda
1, which is
also called
interleukin-29
(IL-29),
binds to a
different
receptor
than IFN
α,
but
downstream
signaling
pathways of
IFN lambda
and IFN
α are
largely
comparable.
The IFN
lambda
receptor is
predominantly
expressed in
hepatocytes.
Thus,
interferon-related
side effects
may be less
frequent
during PEG-IFN
lambda
treatment. A
Phase I
clinical
trial
evaluating
pegylated
interferon
lambda with
or without
ribavirin
was
completed
(Muir 2010).
Interferon
lambda was
well-tolerated
and the
majority of
patients
achieved a
greater than
2 log10
decline of
HCV RNA by 4
weeks.
According to
an interim
analysis of
a subsequent
Phase II
clinical
trial, PEG-IFN
lambda (240
ug, 180 ug,
or 120 ug
once weekly)
was compared
to PEG-IFN
α-2a. PEG-IFN
lambda at
doses of 240
or 180 ug
resulted in
approximately
10% higher
RVR and
approximately
20% higher
cEVR rates,
a lower
frequency of
flu-like
symptoms,
but with
more
frequent
aminotransferase
and
bilirubin
elevations
than PEG-IFN
α-2a (Zeuzem
2011).
Telaprevir-
and
boceprevir-based
triple
therapy of
treatment-naïve
and
treatment-experienced
HCV genotype
1 patients
results in
substantially
increased
SVR rates
compared to
PEG-INF-α
and
ribavirin
alone. The
approval of
these agents
represents a
major
breakthrough
in the
treatment of
chronic
hepatitis C.
However,
successful
use of these
drugs will
require a
precise
classification
of response
patterns to
previous
treatment,
careful
on-treatment
monitoring
of HCV viral
load and
emergence of
antiviral
resistance
as well as
of
additional
side effects
and numerous
possible
drug-drug
interactions.
Next-generation
NS3-4A
protease
inhibitors
and NS5A
inhibitors
may have
even more
favorable
properties
than
telaprevir
and
boceprevir
in terms of
HCV genotype
coverage,
safety
profiles,
less
pronounced
drug-drug
interactions,
or possible
once-daily
administration.
However, the
triple
therapy
approach has
several
limitations.
First of
all,
concomitant
IFN α and
ribavirin
are
necessary to
avoid the
development
of antiviral
resistance.
Consequently,
the efficacy
of triple
therapy was
limited in
prior null
responders
to PEG-IFN α
and
ribavirin,
and triple
therapy
cannot be
administered
to patients
with
contraindications
to PEG-IFN α
or
ribavirin.
Recent data
indicate
that the
development
of DAA
combination
therapies in
all-oral or
quadruple
treatment
regimens
will likely
be a very
potent
option for
these
patients.
In such DAA
combination
regimens,
the
inclusion of
drugs with a
high genetic
barrier to
resistance
such as
nucleoside
NS5B
inhibitors
or drugs
targeting
host factors
such as
alisporivir
may be
important.
Afdhal N,
O'Brien C,
Godofsky E.
Valopicitabine
alone or
with
PEG-interferon/ribavirin
retreatment
in patients
with HCV-1
infection
and prior
non-reponse
to PEGIFN/RBV:
One-year
results. J
Hepatol
2007;46:5.
Ahmed-Belkacem
A, Ahnou N,
Barbotte L,
et al.
Silibinin
and related
compounds
are direct
inhibitors
of hepatitis
C virus
RNA-dependent
RNA
polymerase.
Gastroenterology
2010;138:1112-22.
(Abstract)
Ali S,
Leveque V,
Le Pogam S,
et al.
Selected
replicon
variants
with
low-level in
vitro
resistance
to the
hepatitis C
virus NS5B
polymerase
inhibitor
PSI-6130
lack
cross-resistance
with R1479.
Antimicrob
Agents
Chemother
2008;52:4356-69.
(Abstract)
Bacon BR,
Gordon SC,
Lawitz E, et
al.
Boceprevir
for
previously
treated
chronic HCV
genotype 1
infection. N
Engl J Med
2011;364:1207-17.
(Abstract)
Barnard RJ,
Zeuzem S,
Vierling J,
Sulkowski M,
Manns M,
Long J.
Analysis of
resistance-associated
amino acid
variants in
non-SVR
patients
enrolled in
a
retrospective
long-term
follow-up
analysis of
boceprevir
phase 3
clinical
trials.
Hepatology
2011;54:164.
Bartenschlager
R, Frese M,
Pietschmann
T. Novel
insights
into
hepatitis C
virus
replication
and
persistence.
Adv Virus
Res
2004;63:71-180.
(Abstract)
Beaulieu PL.
Non-nucleoside
inhibitors
of the HCV
NS5B
polymerase:
progress in
the
discovery
and
development
of novel
agents for
the
treatment of
HCV
infections.
Curr Opin
Investig
Drugs
2007;8:614-34.
(Abstract)
Brainard DM,
Petry A, Van
Dyck K, et
al. Safety
and
antiviral
activity of
MK-5172, a
novel HCV
NS3/4A
protease
inhibitor
with potent
activity
against
known
resistance
mutants, in
genotype 1
and 3
HCV-infected
patients.
Hepatology
2010;52:706.
Chayama K,
Takahashi S,
Toyota J, et
al. Dual
therapy with
the NS5A
inhibitor
BMS-790052
and the NS3
protease
inhibitor
BMS-650032
in HCV
genotype
1b-infected
null
responders.
Hepatology
2011, in
press. (Abstract)
Cooper C,
Lawitz E,
Ghali P, et
al.
Antiviral
activity of
the
non-nucleoside
polymerase
inhibitor,
VCH-759, in
chronic
hepatitis C
patients:
Results from
a
randomized,double-blind,
placebo-controlled,
ascending
multiple
dose study.
Hepatology
2007;46:864.
Davis GL,
Nelson DR,
Terrault N,
et al. A
randomized,
open-label
study to
evaluate the
safety and
pharmacokinetics
of human
hepatitis C
immune
globulin (Civacir)
in liver
transplant
recipients.
Liver
Transpl
2005;11:941-9.
(Abstract)
Erhardt A,
Deterding K,
Benhamou Y,
et al.
Safety,
pharmacokinetics
and
antiviral
effect of
BILB 1941, a
novel
hepatitis C
virus RNA
polymerase
inhibitor,
after 5 days
oral
treatment.
Antivir Ther
2009;14:23-32.
(Abstract)
Ferenci P,
Scherzer TM,
Kerschner H,
et al.
Silibinin is
a potent
antiviral
agent in
patients
with chronic
hepatitis C
not
responding
to pegylated
interferon/ribavirin
therapy.
Gastroenterology
2008;135:1561-7.
(Abstract)
Flisiak R,
Feinman SV,
Jablkowski
M, et al.
The
cyclophilin
inhibitor
Debio 025
combined
with PEG IFN
alpha 2a
significantly
reduces
viral load
in
treatment-naive
hepatitis C
patients.
Hepatology
2009;49:1460-8.
(Abstract)
Flisiak R,
Horban A,
Gallay P, et
al. The
cyclophilin
inhibitor
Debio-025
shows potent
anti-hepatitis
C effect in
patients
coinfected
with
hepatitis C
and human
immunodeficiency
virus.
Hepatology
2008;47:817-26.
(Abstract)
Flisiak R,
Pawlotsky JM,
Crabbe R,
Callistru
PI, Kryczka
W,
Häussinger
D. Once
daily
alisporivir
(Debio025)
plus
pegIFNalfa2a/ribavirin
results in
superior
sustained
virologic
response
(SVR) in
chronic
hepatitis C
genotype 1
treatment
naive
patients. J
Hepatol
2011;54:24.
Fridell RA,
Wang C, Sun
JH, et al.
Genotypic
and
phenotypic
analysis of
variants
resistant to
hepatitis C
virus
nonstructural
protein 5A
replication
complex
inhibitor
BMS-790052
in Humans:
In Vitro and
In Vivo
Correlations.
Hepatology
2011, in
press. (Abstract)
Gane EJ,
Roberts SK,
Stedman CA,
et al. Oral
combination
therapy with
a nucleoside
polymerase
inhibitor
(RG7128) and
danoprevir
for chronic
hepatitis C
genotype 1
infection
(INFORM-1):
a randomised,
double-blind,
placebo-controlled,
dose-escalation
trial.
Lancet
2010;376:1467-75.
(Abstract)
Gane EJ,
Stedman CA,
Hyland RH,
et al. Once
daily
PSI-7977
plus RBV:
pegylated
interferon-alpha
not required
for complete
rapid viral
response in
treatment-naive
patients
with HCV Gt2
or Gt3.
Hepatology
2011;54:18.
Gao M,
Nettles RE,
Belema M, et
al. Chemical
genetics
strategy
identifies
an HCV NS5A
inhibitor
with a
potent
clinical
effect.
Nature
2010;465:96-100.
(Abstract)
Gaudieri S,
Rauch A,
Pfafferott
K, et al.
Hepatitis C
virus drug
resistance
and
immune-driven
adaptations:
relevance to
new
antiviral
therapy.
Hepatology
2009;49:1069-82.
(Abstract)
Hezode C,
Hirschfield
GM,
Ghesquiere
W, et al.
BMS-790052,
a NS5A
replication
complex
inhibitor,
combined
with
peginterferon
alfa-2a and
ribavirin in
treatment-navie
HCV-gentoype
1 or 4
patients:
phase 2b
AI444010
study
interim week
12 results.
Hepatology
2011;54:115.
Hinrichsen
H, Benhamou
Y, Wedemeyer
H, et al.
Short-term
antiviral
efficacy of
BILN 2061, a
hepatitis C
virus serine
protease
inhibitor,
in hepatitis
C genotype 1
patients.
Gastroenterology
2004;127:1347-55.
(Abstract)
Jacobson IM,
McHutchison
JG, Dusheiko
G, et al.
Telaprevir
for
previously
untreated
chronic
hepatitis C
virus
infection. N
Engl J Med
2011;364:2405-16.
(Abstract)
Jacobson IM,
Pockros PJ,
Lalezari JP,
et al.
Virologic
response
rates
following 4
weeks of
filibuvir in
combination
with
pegylated
interferon-alpha-2a
and ribavrin
in
chronically
infected HCV
genotype1
patients. J
Hepatol
2010;52:465.
Janssen HL,
Reesink HW,
Zeuzem S, et
al. A
randomized,
double-blind,
placebo
controlled
safety and
anti-viral
proof of
concept
study of
miravirsen,
an
oligonucleotide
targeting
mir-122, in
treatment
naive
patients
with
gentoype 1
chronic HCV
infection.
Hepatology
2010;54:1071.
Kim JL,
Morgenstern
KA, Griffith
JP, et al.
Hepatitis C
virus NS3
RNA helicase
domain with
a bound
oligonucleotide:
the crystal
structure
provides
insights
into the
mode of
unwinding.
Structure
1998;6:89-100.
(Abstract)
Kim JL,
Morgenstern
KA, Lin C,
et al.
Crystal
structure of
the
hepatitis C
virus NS3
protease
domain
complexed
with a
synthetic
NS4A
cofactor
peptide.
Cell
1996;87:343-55.
(Abstract)
Kneteman NM,
Howe AY, Gao
T, et al.
HCV796: A
selective
nonstructural
protein 5B
polymerase
inhibitor
with potent
anti-hepatitis
C virus
activity in
vitro, in
mice with
chimeric
human
livers, and
in humans
infected
with
hepatitis C
virus.
Hepatology
2009;49:745-52.
(Abstract)
Koch U,
Narjes F.
Allosteric
inhibition
of the
hepatitis C
virus NS5B
RNA
dependent
RNA
polymerase.
Infect
Disord Drug
Targets
2006;6:31-41.
(Abstract)
Lamarre D,
Anderson PC,
Bailey M, et
al. An NS3
protease
inhibitor
with
antiviral
effects in
humans
infected
with
hepatitis C
virus.
Nature
2003;426:186-9.
(Abstract)
Landford RE,
Hildebrandt-Eriksen
ES, Petri A,
et al.
Therapeutic
silencing of
microRNA-122
in primates
with chronic
hepatitis C
virus
infection.
Science
2010;327:198-201.
(Abstract)
Lawitz E,
Lalezari JP,
Hassanein T,
et al.
Once-daily
PSI-7977
plus peg/RBV
in
treatment-naive
pateitns
with HCV
GT1: robust
end of
treatemnt
response
rates are
sustained
post-treatment.
Hepatology
2011;54:113.
Lenz O,
Fevery B,
Vigen L, et
al. TMC435
in
combination
with
peginterferon
alpha-2a/ribavirin
in
treatment-naive
patients
infected
with HCV
genotype 1:
virology
analysis of
the Pillar
study.
Hepatology
2011;54:985.
Lesburg CA,
Cable MB,
Ferrari E,
Hong Z,
Mannarino
AF, Weber
PC. Crystal
structure of
the
RNA-dependent
RNA
polymerase
from
hepatitis C
virus
reveals a
fully
encircled
active site.
Nat Struct
Biol
1999;6:937-43.
(Abstract)
Lin C, Gates
CA, Rao BG,
et al. In
vitro
studies of
cross-resistance
mutations
against two
hepatitis C
virus serine
protease
inhibitors,
VX-950 and
BILN 2061.
J Biol Chem
2005;280:36784-91.
(Abstract)
Lindenbach
BD, Evans
MJ, Syder
AJ, et al.
Complete
replication
of hepatitis
C virus in
cell
culture.
Science
2005;309:623-6.
(Abstract)
Lohmann V,
Korner F,
Koch J,
Herian U,
Theilmann L,
Bartenschlager
R.
Replication
of
subgenomic
hepatitis C
virus RNAs
in a
hepatoma
cell line.
Science
1999;285:110-3.
Lok AS,
Gardiner DF,
Lawitz E.
Quadruple
therapy with
BMS-790052,
BMS-650032
and Peg-IFN/RBV
for 24 weeks
results in
100% SVR12
in HCV
genotype
null
responders.
J Hepatol
2011;54:536.
Lorenz IC,
Marcotrigiano
J, Dentzer
TG, Rice CM.
Structure of
the
catalytic
domain of
the
hepatitis C
virus NS2-3
protease.
Nature
2006;442:831-5.
(Abstract)
Manns M,
Reesink H,
Berg T, et
al.
Rapid viral
response of
once-daily
TMC435 plus
pegylated
interferon/ribavirin
in hepatitis
C genotype-1
patients: a
randomized
trial.
Antivir Ther
2011;16:1021-33.
(Abstract)
McCown MF,
Rajyaguru S,
Kular S,
Cammack N,
Najera I.
GT-1a or
GT-1b
subtype-specific
resistance
profiles for
hepatitis C
virus
inhibitors
telaprevir
and HCV-796.
Antimicrob
Agents
Chemother
2009;53:2129-32.
(Abstract)
Meylan E,
Curran J,
Hofmann K,
et al.
Cardif is an
adaptor
protein in
the RIG-I
antiviral
pathway and
is targeted
by hepatitis
C virus.
Nature
2005;437:1167-72.
(Abstract)
Moradpour D,
Penin F,
Rice CM.
Replication
of hepatitis
C virus. Nat
Rev
Microbiol
2007;5:453-63.
(Abstract)
Muir AJ,
Shiffman ML,
Zaman A, et
al. Phase 1b
study of
pegylated
interferon
lambda 1
with or
without
ribavirin in
patients
with chronic
genotype 1
hepatitis C
virus
infection.
Hepatology
2010;52:822-32.
(Abstract)
Nelson DR,
Gane EJ,
Jacobson IM,
Di Bisceglie
AM, Alves K,
Koziel MJ.
VX-222/Telaprevir
in
combination
with
peginterferon-alfa-2a
and
ribavirin in
treatment-naive
genotype 1
HCV patients
treated for
12 weeks:
Zenith
study, SVR
12 interim
analyses.
Hepatology
2011;54:1442.
Nettles RE,
Gao M,
Bifano M, et
al. Multiple
ascending
dose study
of
BMS-790052,
an NS5A
replication
complex
inhibitor,
in patients
infected
with
hepatitis C
virus
genotype 1.
Hepatology
2011, in
press.
Pawlotsky JM,
Flisiak R,
Rasenack J,
et al. Once
daily
alisporivir
interferon-free
regimens
achieve high
rates of
early HCV
clearance in
previously
untreated
patients
with HCV
gentoype 2
or 3.
Hepatology
2011;54:1074.
Pockros P,
Jensen DM,
Tsai N.
First SVR
data with
the
nucleoside
analogue
polymerase
inhibitor
mericitabine
(RG7128)
combined
with
peginterferon/ribavirin
in
treatment-naive
HCV G1/4
patients:
interim
analysis of
the JUMP-C
trial. J
Hepatol
2011;54:538.
Pockros P,
Nelson D,
Godofsky E,
et al. High
relapse rate
seen at week
72 for
patients
treated with
R1626
combination
therapy.
Hepatology
2008;48:1349-50.
(Abstract)
Pockros PJ,
Nelson D,
Godofsky E,
et al. R1626
plus
peginterferon
Alfa-2a
provides
potent
suppression
of hepatitis
C virus RNA
and
significant
antiviral
synergy in
combination
with
ribavirin.
Hepatology
2008;48:385-97.
(Abstract)
Poordad F,
McCone J Jr,
Bacon BR, et
al.
Boceprevir
for
untreated
chronic HCV
genotype 1
infection. N
Engl J Med
2011;364:1195-206.
(Abstract)
Reesink HW,
Fanning GC,
Farha KA, et
al. Rapid
HCV-RNA
decline with
once daily
TMC435: a
phase I
study in
healthy
volunteers
and
hepatitis C
patients.
Gastroenterology
2010;138:913-21.
(Abstract)
Reesink HW,
Zeuzem S,
Weegink CJ,
et al. Rapid
decline of
viral RNA in
hepatitis C
patients
treated with
VX-950: a
phase Ib,
placebo-controlled,
randomized
study.
Gastroenterology
2006;131:997-1002.
(Abstract)
Reiser M,
Hinrichsen
H, Benhamou
Y, et al.
Antiviral
efficacy of
NS3-serine
protease
inhibitor
BILN-2061 in
patients
with chronic
genotype 2
and 3
hepatitis C.
Hepatology
2005;41:832-5.
(Abstract)
Rossignol JF,
Elfert A,
El-Gohary Y,
Keeffe EB.
Improved
virologic
response in
chronic
hepatitis C
genotype 4
treated with
nitazoxanide,
peginterferon,
and
ribavirin.
Gastroenterology
2009;136:856-62.
(Abstract)
Sarrazin C,
Kieffer TL,
Bartels D,
et al.
Dynamic
hepatitis C
virus
genotypic
and
phenotypic
changes in
patients
treated with
the protease
inhibitor
telaprevir.
Gastroenterology
2007;132:1767-77.
(Abstract)
Sarrazin C,
Rouzier R,
Wagner F, et
al. SCH
503034, a
novel
hepatitis C
virus
protease
inhibitor,
plus
pegylated
interferon
alpha-2b for
genotype 1
nonresponders.
Gastroenterology
2007;132:1270-8.
(Abstract)
Sarrazin C,
Zeuzem S.
Resistance
to direct
antiviral
agents in
patients
with
hepatitis C
virus
infection.
Gastroenterology
2010;138:447-62.
(Abstract)
Schiano TD,
Charlton M,
Younossi Z,
et al.
Monoclonal
antibody
HCV-AbXTL68
in patients
undergoing
liver
transplantation
for HCV:
results of a
phase 2
randomized
study. Liver
Transpl
2006;12:1381-9.
(Abstract)
Sherman KE,
Flamm SL,
Afdhal NH,
et al.
Response-guided
telaprevir
combination
treatment
for
hepatitis C
virus
infection. N
Engl J Med
2011;365:1014-24.
(Abstract)
Sherman KE,
Sulkowski M,
Zoulim F,
Alberti A.
Follow-up of
SVR
durability
and viral
resistance
in patients
with chronic
hepatitis C
treatetd
with
telaprevir-based
regimens:
interim
analysis of
the extend
study.
Hepatology
2011;54:1471.
Shi ST,
Polyak SJ,
Tu H, Taylor
DR, Gretch
DR, Lai MM.
Hepatitis C
virus NS5A
colocalizes
with the
core protein
on lipid
droplets and
interacts
with
apolipoproteins.
Virology
2002;292:198-210.
(Abstract)
Susser S,
Vermehren J,
Forestier N,
et al.
Analysis of
long-term
persistence
of
resistance
mutations
within the
hepatitis C
virus NS3
protease
after
treatment
with
telaprevir
or
boceprevir.
J Clin Virol
2011. (Abstract)
Tellinghuisen
TL,
Marcotrigiano
J, Rice CM.
Structure of
the
zinc-binding
domain of an
essential
component of
the
hepatitis C
virus
replicase.
Nature
2005;435:374-9.
(Abstract)
Wagoner J,
Negash A,
Kane OJ, et
al. Multiple
effects of
silymarin on
the
hepatitis C
virus
lifecycle.
Hepatology
2010;51:1912-21.
(Abstract)
Wakita T,
Pietschmann
T, Kato T,
et al.
Production
of
infectious
hepatitis C
virus in
tissue
culture from
a cloned
viral
genome. Nat
Med
2005;11:791-6.
(Abstract)
Welsch C,
Domingues
FS, Susser
S, et al.
Molecular
basis of
telaprevir
resistance
due to V36
and T54
mutations in
the NS3-4A
protease of
the
hepatitis C
virus.
Genome Biol
2008;9:R16.
(Abstract)
Wohnsland A,
Hofmann WP,
Sarrazin C.
Viral
determinants
of
resistance
to treatment
in patients
with
hepatitis C.
Clin
Microbiol
Rev
2007;20:23-38.
(Abstract)
Zeuzem S,
Andreone P,
Pol S, et
al.
Telaprevir
for
retreatment
of HCV
infection. N
Engl J Med
2011;364:2417-28.
(Abstract)
Zeuzem S,
Arora S,
Bacon B, Box
T, Charlton
M. Pegylated
interferon-lambda
shows
superior
viral
response
with
improved
safety and
tolerability
versus
pegIFN-alfa-2a
in hCV
patients
(G1/2/3/4):
merge phase
IIB trhough
week 12. J
Hepatol
2011;54:538.
Zeuzem S,
Asselah T,
Angus P, et
al. Efficacy
of the
Protease
Inhibitor BI
201335,
Polymerase
Inhibitor BI
207127, and
Ribavirin in
Patients
With Chronic
HCV
Infection.
Gastroenterology
2011, in
press. (Abstract)
Zeuzem S,
Berg T,
Moeller B,
et al.
Expert
opinion on
the
treatment of
patients
with chronic
hepatitis C.
J Viral
Hepat
2009;16:75-90.
(Abstract)
Zeuzem S,
Buggisch P,
Agarwal K,
et al. The
protease
inhibitor
GS-9256 and
non-nucleoside
polymerase
inhibitor
tegobuvir
alone, with
RBV or
peginterferon
plus RBV in
hepatitis C.
Hepatology
2011, in
press. (Abstract)
Zeuzem S,
Sulkowski
MS, Lawitz
EJ, et al.
Albinterferon
Alfa-2b was
not inferior
to pegylated
interferon-alpha
in a
randomized
trial of
patients
with chronic
hepatitis C
virus
genotype 1.
Gastroenterology
2010;139:1257-66.
(Abstract)